Klicken Sie auf eine Abbildung, um dass nächste Bild zu sehen. Alternativ können Sie auf die schwarzen Punkte klicken, um vorwärts und zurück zu navigieren.
Einleitung
▼ ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ●
Abb. 1: Der QR-Code verweist auf die Literaturliste zu diesem Buch.
● ● ▼ ● ●
Abb. 2: Schematische Illustration des Cas9-Proteins der Genschere.
● ● ● ▼ ●
Abb. 3: Die Goldene Schallplatte. Bilder: NASA.
● ● ● ● ▼
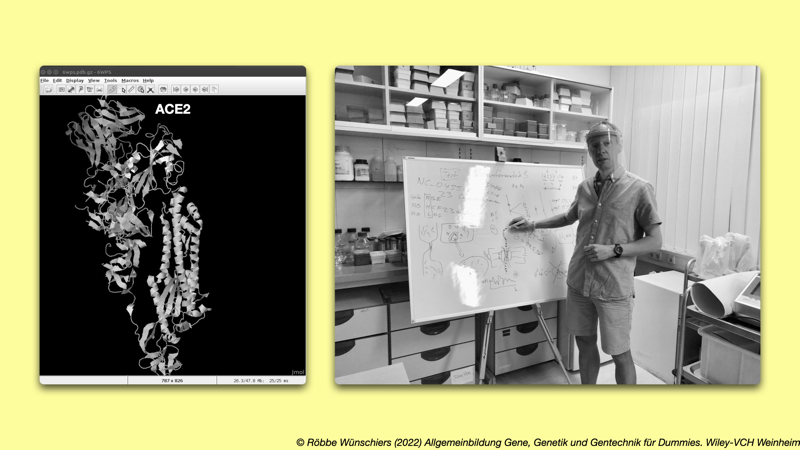
Abb. 4: Unterricht und Diskussion über Coronaviren mit Schülern. Foto: Heidi Fußwinkel. Kapitel 1: Die Welt der Gene
▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Abb. 1.1: Modell der DNA-Doppelhelix. Diese Zeichnung aus der Veröffentlichung von James Watson und Francis Crick hat Cricks Frau Odile Crick gezeichnet. Sie verwendete dabei mutmaßlich die Proportionen des Goldenen Schnitts. Die der Struktur zugrundeliegenden Daten wurden von Rosalind Franklin per Röntgenstrukturanalyse gewonnen. Sie starb, bevor der Nobelpreis 1962 für die Strukturaufklärung vergeben wurde.
● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
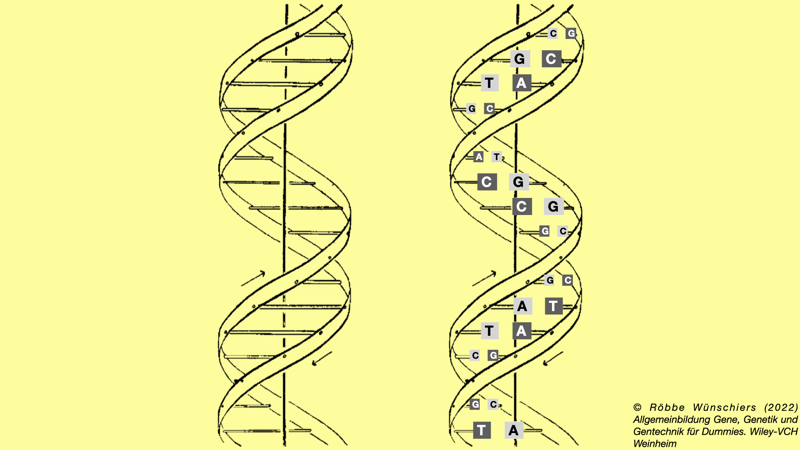
Abb. 1.2: Schematische Darstellung der DNA. Der obere Strang ist komplementär zu dem Gegenstrang, da immer A und T sowie C und G gepaart sind.
● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ●
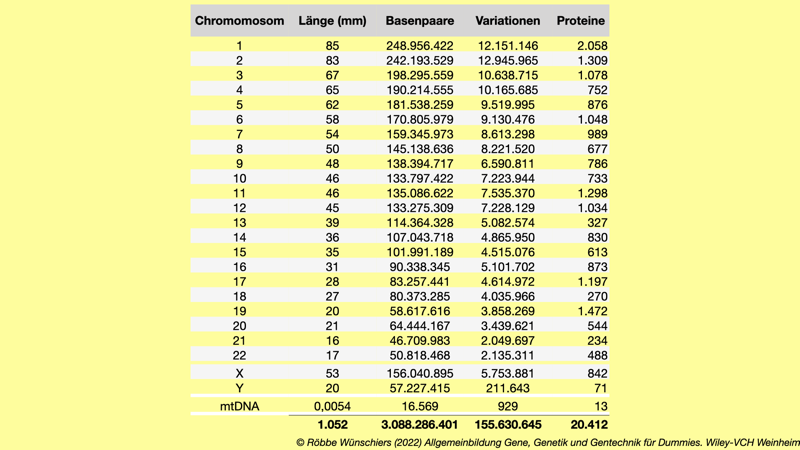
Tab. 1.1: Ausgewählte Daten zu den Chromosomen eines Mannes. Das weibliche Erbgut ist mit 3.187.099.881 Basenpaaren rund hunderttausend Basenpaare größer.
● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ●
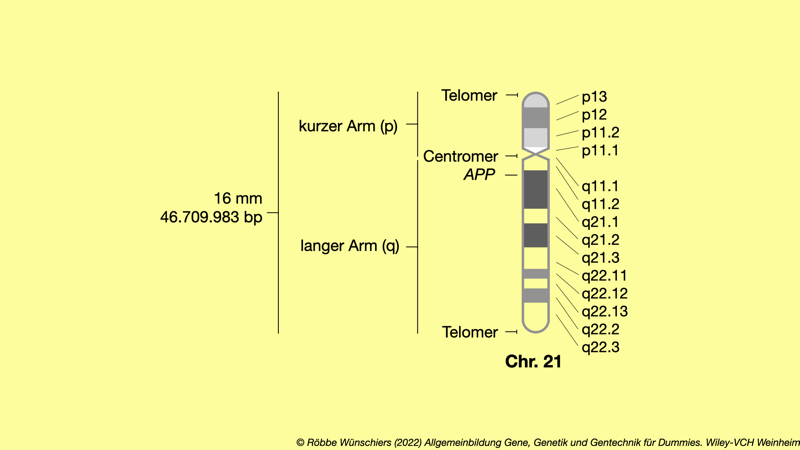
Abb. 1.3: Idiogramm des Chromosoms 21, auf dem auch das Gen APP liegt. Das Gen APP an Position 21q21.3 (Basenpaare 25.880.550 bis 26.171.128) codiert für das Amyloid Precursor Protein, das an der Alzheimererkrankung beteiligt ist. Nach seiner Auflösung (Proteolyse) bildet es einen Hauptbestandteil der Plaques, die in den Gehirnen von Alzheimer-Patienten gefunden werden.
● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ●
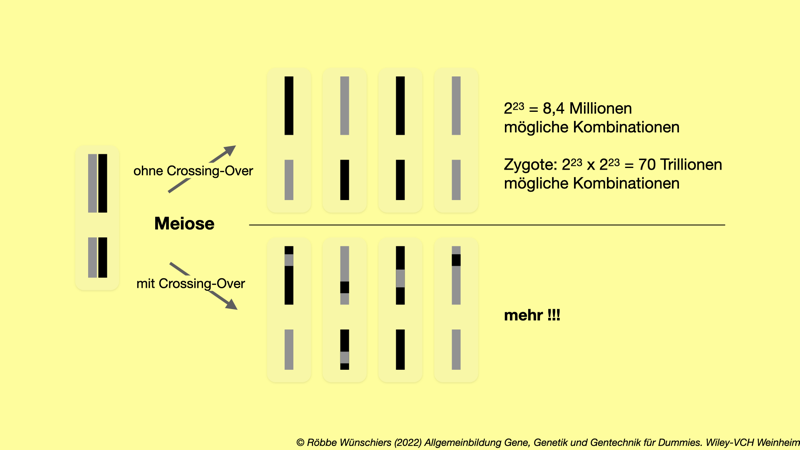
Abb. 1.4: Die Meiose führt zur Entstehung von Samen und Eizellen. Dazu wird der doppelte (diploide) Chromosomensatz in zwei einfache (haploide) Chromosomensätze aufgeteilt. Im Beispiel sind nur zwei Chromosomen gezeigt. Bei 23 Chromosomen gibt es 8,4 Millionen Kombinationsmöglichkeiten. Bei der anschließenden Befruchtung mit Keimzellen eines anderen Organismus quadriert sich die Zahl der möglichen Kombinationen. Findet zusätzlich noch Rekombination in Form von Crossing-Over statt, werden einzelne Abschnitte der Schwesterchromosomen ausgetauscht. Die Anzahl der Kombinationen – ganz ohne Mutationen – ist unermesslich. Es lebe der Sex.
● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ●
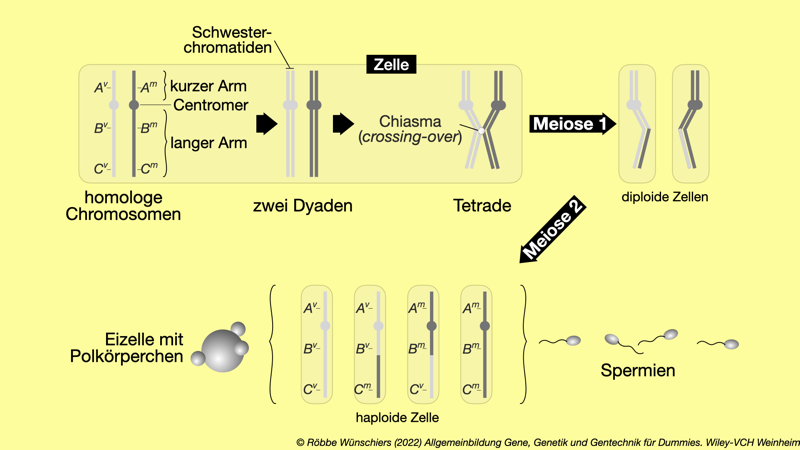
Abb. 1.5: Tanz der Chromosomen während der Meiose am Beispiel eines einzelnen Chromosomenpaares. Auf den homologen Chromosomen, je ein mütterliches und ein väterliches, habe ich die Position von drei Genen A, B, C) markiert. Das väterliche Allel ist mit v und mütterliche Allel mit m gekennzeichnet. Während der Meiose 1 wird das Chromosomenpaar verdoppelt. Es entsteht je eine Dyade (griechisch für Zweiheit) mit zwei Schwesterchromatiden. Jedes Chromosom bekommet also ein Schwesterchen. Diese Dyaden bilden daraufhin eine Tetrade (griechische Doppelkopfrunde – Scherz), bei der sich Nichtschwesterchromatide (quasi Cousinenchromatide) aneinander anlagern und dabei ein sogenanntes Chiasma (griechisch für Kreuzung; benannt nach dem griechischen Buchstaben Chi) bilden können. Hier kommt es dann zum Crossing-Over, also dem Austausch zwischen den beiden DNA-Strängen. Während der Meiose 2 werden alle Chromatiden voneinander getrennt und auf einzelne Zellen, die Keimzellen, verteilt.
● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ●
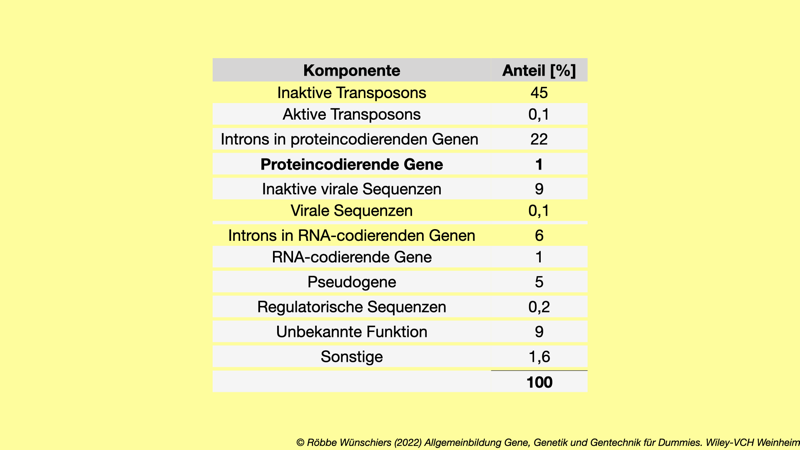
Tab. 1.2: Anteile funktionaler Komponenten, die in unserem Genom codiert sind.
● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ●
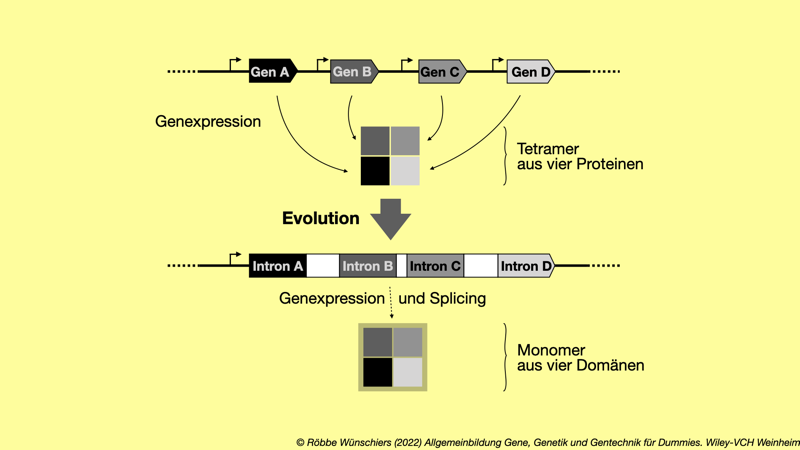
Abb. 1.6: Die Exontheorie der Gene. Die Theorie besagt, dass früh in der Evolution kleine Proteine mit einer einzelnen Funktion vorlagen. Im Laufe der Evolution sind diese Gene zu einem Gen zusammengefügt worden, das ein einzelnes Protein mit mehreren funktionellen Einheiten (Domänen) codiert.
● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ●
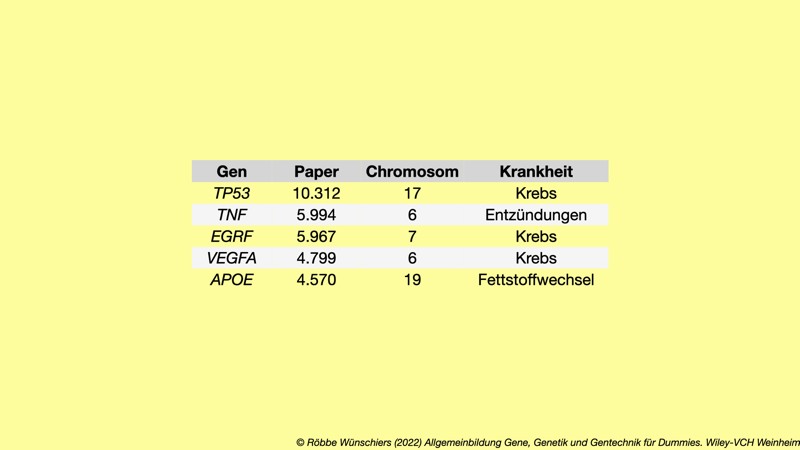
Tab. 1.3: Gene des Menschen mit den meisten Erwähnungen in der wissenschaftlichen Primärliteratur (im Wissenschaftsjargon Papern) und deren wichtigste Beteiligung an Krankheiten. Stand: 02. Juni 2021.
● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ●
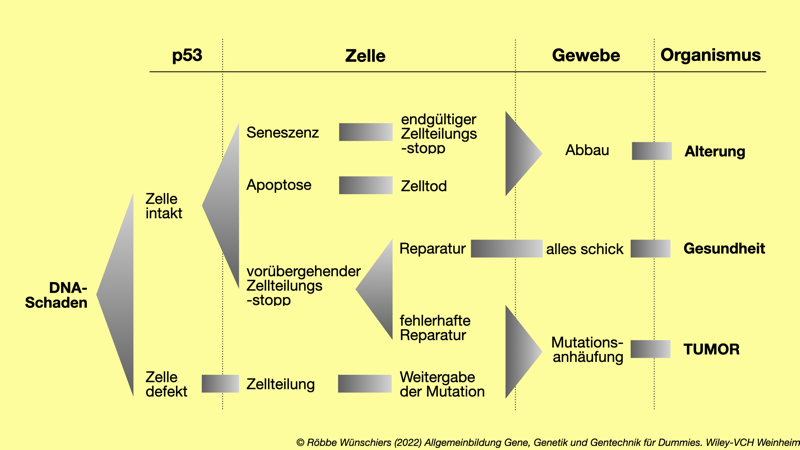
Abb. 1.7: Wirkung des »Genomwächters« p53 auf zellbiologische Prozesse, die über Leben und Tod einer Zelle und ihres Gewebes entscheiden.
● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ●
Abb. 1.8: Ein einzelner Unterschied in der Abfolge der Nukleotide, also der Gensequenz, reicht, um Allele voneinander zu unterscheiden.
● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ●
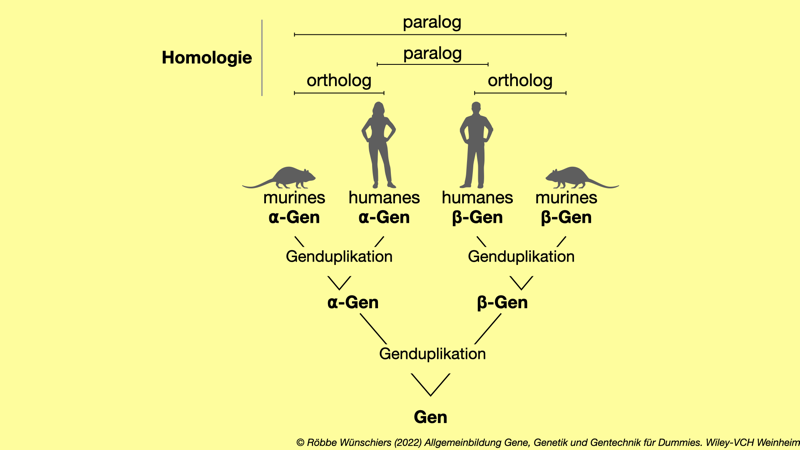
Abb. 1.9: Konzept der Homologie von Genen und Proteinen. Homologe Gene stammen von demselben »Urgen« ab. Befinden sich die Duplikate in dem haploiden Erbgut einer Art, so spricht man von paralogen Genen oder Proteinen. Homologe Gene und Proteine bei unterschiedlichen Arten werden dagegen als ortholog bezeichnet.
● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ●
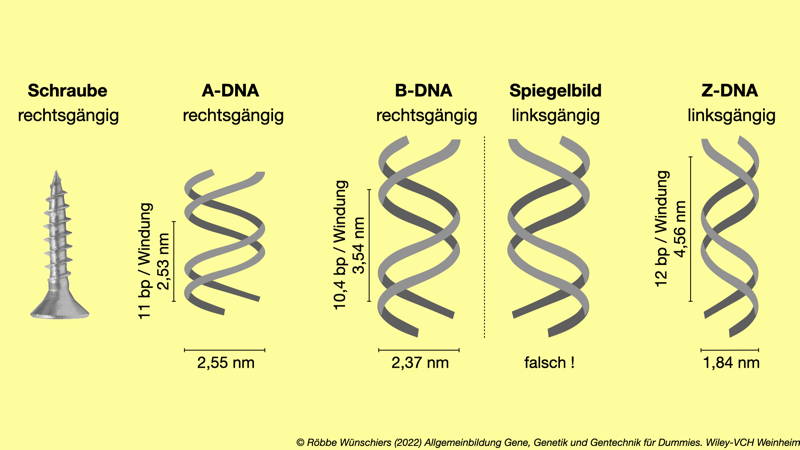
Abb. 1.10: Strukturvarianten der DNA, die unter anderem eine Rolle bei der Regulation der Genaktivität spielen. Die B-DNA ist jene von James Watson und Francis Crick beschriebene Form. Wie eine Schraube mit rechtssteigendem Gewinde ist die Drehrichtung der A- und B-DNA im Uhrzeigersinn. Die Zickzackform der Z-DNA entspricht einem Linksgewinde.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ●
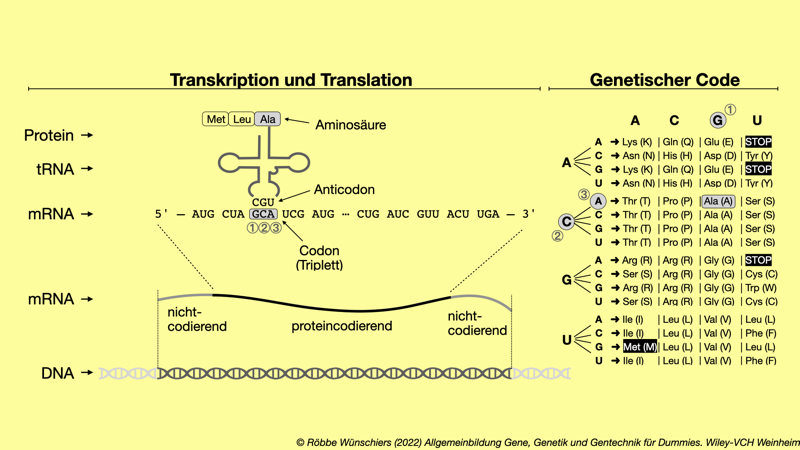
Abb. 1.11: Der genetische Code. Einzelne Aminosäuren, die Bausteine der Proteine, werden durch Codons (Tripletts) codiert. Im universellen genetischen Code auf der rechten Seite sind die Aminosäuren in ihrer Dreibuchstaben- und, in Klammern, Einbuchstabenankürzung aufgeschrieben. Das Startcodon AUG und die Stopcodons UGA, UAA und UGA sind schwarz unterlegt.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ●
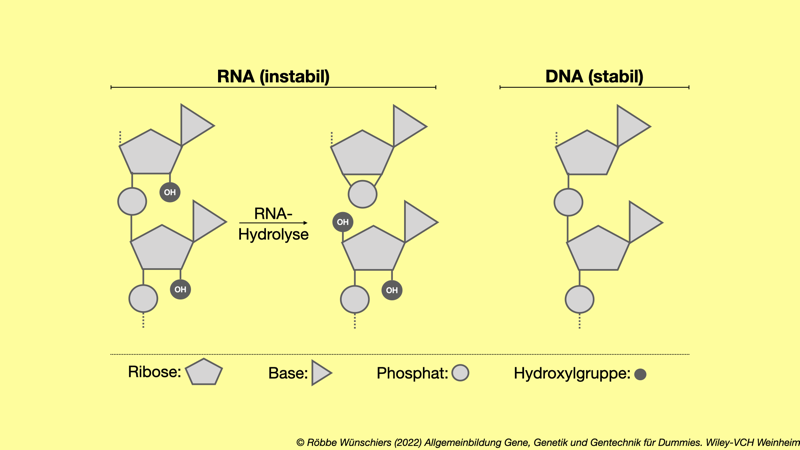
Abb. 1.12: RNA ist instabil und wird schnell abgebaut. Das liegt an den Hydroxlygruppen (-OH) an der Ribose (R). Diese ist empfindlich gegenüber chemischen Attacken, sogenannten nukleophilen Angriffen, die zur Hydrolyse führen. B: Nukleobase; P: Phosphat.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ●
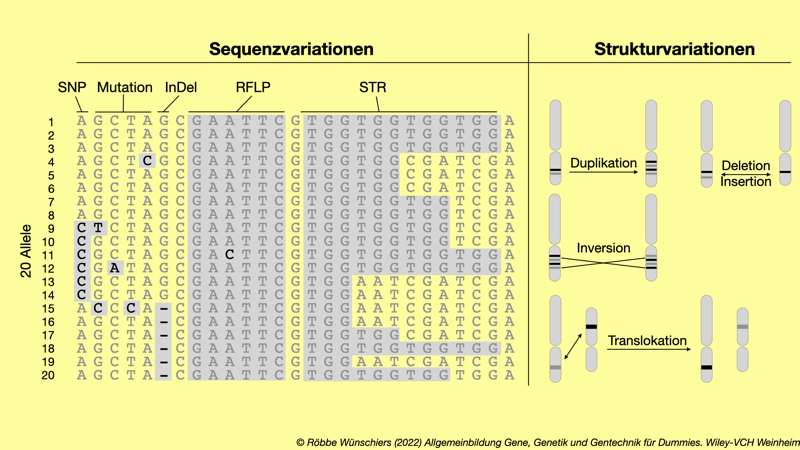
Abb. 1.13: DNA-Sequenz- und Strukturpolymorphismen. Links: Es ist von zwanzig Allelen jeweils nur ein DNA-Strang gezeigt. Einzelnukleotidvariationen (SNPs, englisch: single nucleotide polymorphisms) unterscheiden sich von Mutationen dadurch, dass sie in mindestens einem Prozent der Allele auftreten. Bei Insertionen und Deletionen (InDels, englisch: insertions/deletions) fehlen in einigen Allelen Basenpaare. Eine Mutation A zu C im Allel 11) in der Erkennungssequenz für das Restriktionsenzym EcoRI (GAATTC) bewirkt einen Restriktionspolymorphismus (RFLP, englisch: restriction fragment length polymorphism). Die Mikrosatellitensequenz TGG führt durch unterschiedlich häufige Wiederholungen (STR, englisch: short tandem repeat) zu einem Längenpolymorphismus. Rechts: Es sind verschiedene Polymorphismen auf Chromosomenebene dargestellt.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼
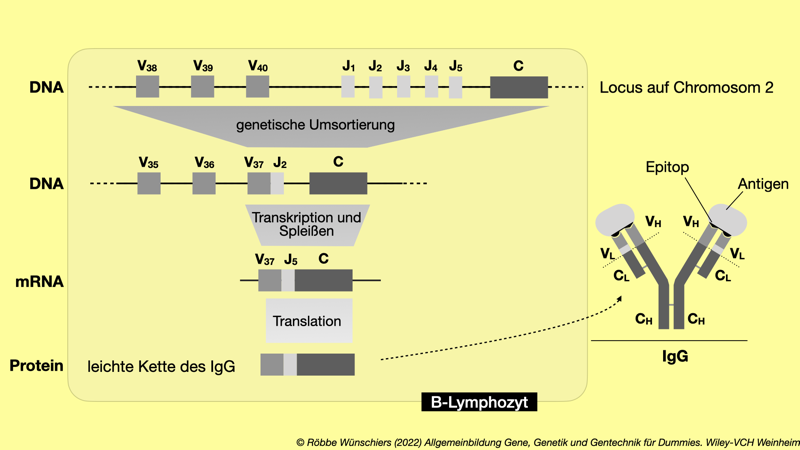
Abb. 1.14: Herstellung und Aufbau eines IgG-Antikörpers. Während der Entwicklung der B-Lymphozyten werden aus dem Immunoglobin-Genlocus je ein V- und ein J-Segment mit einem C-Segment zu einem Gen verbunden. Allein hierbei sind vierzig mal fünf Kombinationen möglich – dadurch das diese Rekombination fehlerhaft sein kann, aber noch mehr. Der ins Blut entlassene IgG-Antikörper besteht aus je zwei identischen schweren und leichten Proteinketten, die miteinander verbunden sind. Deren variable Bereiche erkennen spezifisch einen Bereich (Epitop) eines Fremdmoleküls (Antigen). V: variabel; J: join; C: konstant; L: leicht; H: schwer. Kapitel 2: Von der Vererbung zur quantitativen Genetik
▼ ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ●
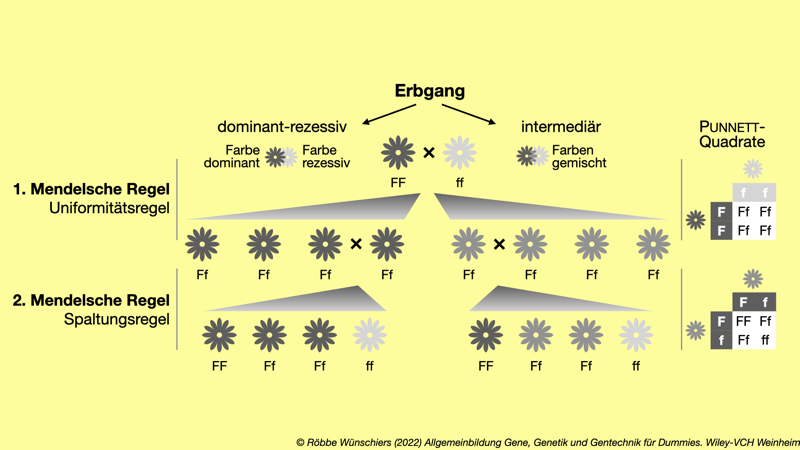
Abb. 2.1: Die Mendelschen Regeln gelten für Merkmale, die von genau einem Gen codiert werden, das in zwei Kopien – also als mütterliches und väterliches Allel – vorliegt. Die erste und zweite Mendelsche Regel beschreiben die Weitergabe der Gene und die Merkmalsausprägung in der ersten und zweiten Tochtergeneration. Das Punnett-Quadrat, benannt nach dem britischen Genetiker Reginald Punnett, beschreibt die möglichen diploiden Genotypen, die aus den haploiden Keimzellen (Allel F und Allel f) entstehen können.
● ● ▼ ● ● ● ● ● ●
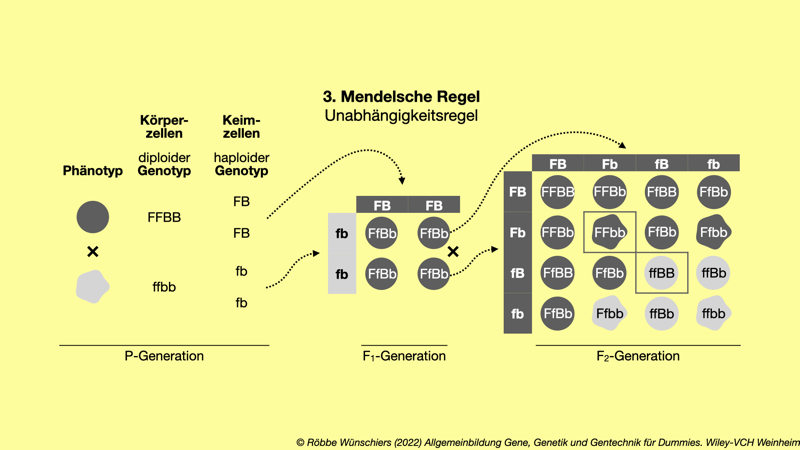
Abb. 2.2: Die dritte Mendelsche Regel beschreibt die unabhängige Vererbung mehrerer Merkmale, wenn diese nicht gekoppelt sind, das heißt, wenn sie auf unterschiedlichen Chromosomen liegen oder auf einem Chromosom weit voneinander entfernt sind. In diesem Beispiel folgen die Merkmale Farbe (F) und Beschaffenheit (B) der Erbsenhülle einem dominat-rezessiven Erbgang. Die Punnett-Quadrate zeigen die aus der Fusion der haploiden Keimzellen möglichen diploiden Genotypen. Die eingerahmten Merkmalskombinationen der F2-Generation kommen nicht in der Elterngeneration vor. Sie sind reinerbige neue Kombinationen.
● ● ● ▼ ● ● ● ● ●
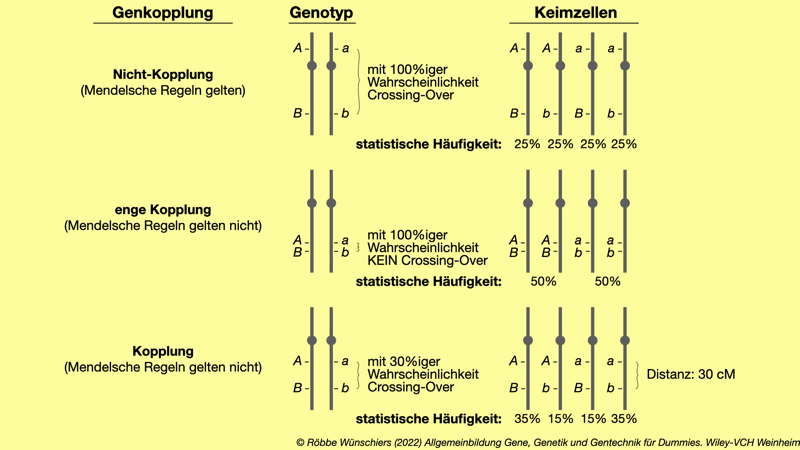
Abb. 2.3: Durch die Kopplung von Genen werden die Mendelschen Regeln außer Kraft gesetzt. Die Rekombinationshäufigkeit (auch Rekombinationsfrequenz genannt) gibt an, wie groß die Wahrscheinlichkeit ist, dass zwischen zwei Orten (Loci) im Genom Crossing-Over stattfindet. Diese Rekombinationswahrscheinlichkeit wurde früher in der Einheit Centimorgan (cM) angegeben.
● ● ● ● ▼ ● ● ● ●
Abb. 2.4: Originalabbildung aus dem Buch Experimentelle Protistenstudien von Victor Jollos von 1921. Die Versuche führte er bereits zehn Jahre vorher durch. Die Selektion der Körpergröße bei Pantoffeltierchen führt in jeder Weiterzucht wieder zu einer Normalverteilung der Körpergrößen wie in der ursprünglichen Population. Das ist das Ergebnis der Regression zum Mittelwert.
● ● ● ● ● ▼ ● ● ●
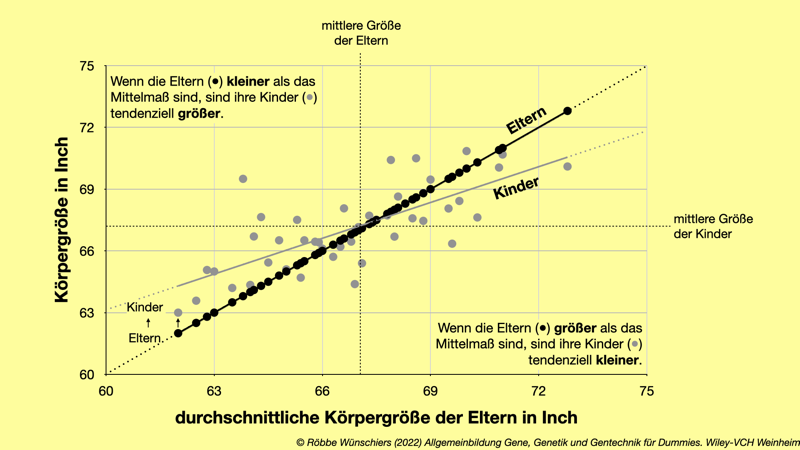
Abb. 2.5: Francis Galton beschrieb 1886 die Regression zum Mittelwert. Schwarze Kreise stellen die mittlere Größe der Elternpaare dar. Graue Kreise kennzeichnen Mittelwerte der Körpergrößen der Kinder. Die Kinder von Eltern, die kleiner als die Durchschnittsgröße sind, tendieren zum Mittelwert, werden also eher größer als die Eltern. Die Umrechnung der Körpergröße von Inch zu Zentimetern macht leider keinen Sinn: Das Inch wurde erst in den 1950er Jahren als 2,54 Zentimetern festgelegt. Vorher gab es Wildwuchs.
● ● ● ● ● ● ▼ ● ●
Abb. 2.6: Die Einflüsse des Erbguts und der Umwelt auf Merkmale (Phänotypen) unterscheiden sich.
● ● ● ● ● ● ● ▼ ●
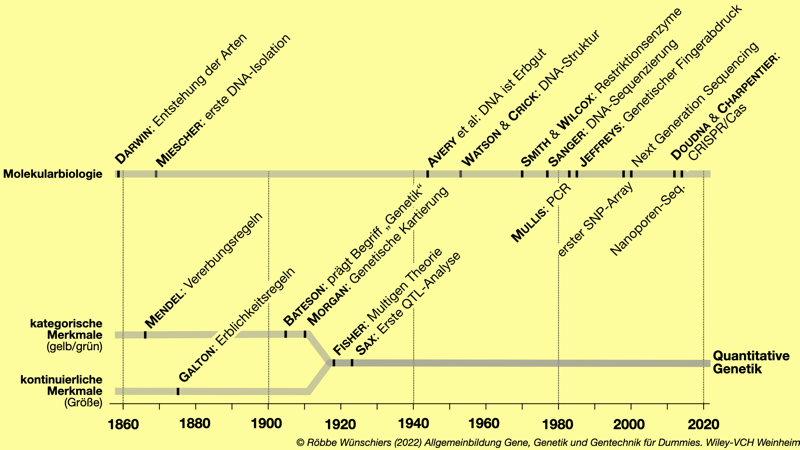
Abb. 2.7: Die quantitative Genetik vereint die Konzepte der Vererbung kategorischer Merkmale nach Mendel und kontinuierlicher Merkmale nach Galton. Vater der Synthese ist der britische Mathematiker Ronald Fisher. Zur Erinnerung: Darwins Werk Über die Entstehung der Arten erschien 1859.
● ● ● ● ● ● ● ● ▼
Abb. 2.8: Ein Überblick über die Blattrundung, die Einkerbungstiefe und die Blattfläche bei der Säulen-Espe (Populus tremula erecta). Jedes Blatt stammt von einem eigenen Genotyp. Ich zeige hier nur eine Auswahl der auftretenden Formvariationen. Kapitel 3: Vom Gen übers Genom zum Ich
▼ ● ● ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ● ● ●
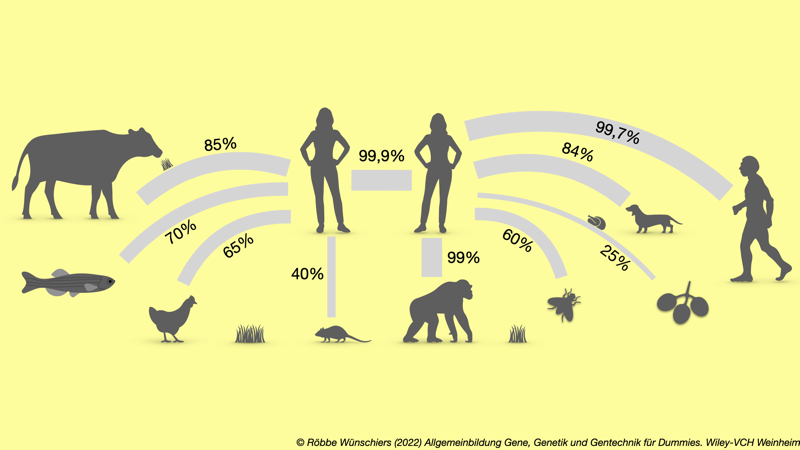
Abb. 3.1: Grad der Übereinstimmung der Genome verschiedener Organismen. Auf der Ebene der Proteine ist die Identität noch viel größer. Der Mensch ganz rechts ist der Neanderthaler. Der Maus, Zebrafisch und Fruchtfliege sind beliebte Modellorganisimen für Krankheiten beim Menschen, da sie sich gut züchten und halten lassen. Schon gemerkt?: Die Größenverhältnisse stimmen nicht.
● ● ▼ ● ● ● ● ● ● ● ●
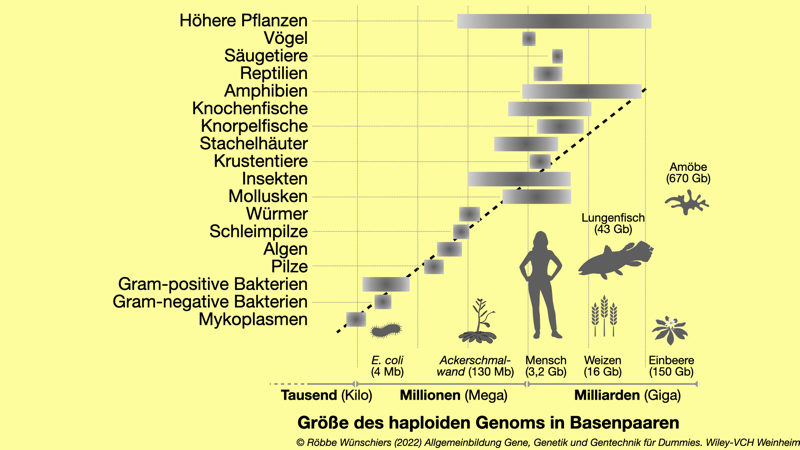
Abb. 3.2: Die Größe von Genomen und das C-Wert Paradox. Die Größe des Erbguts nimmt nicht proportional mit der Komplexität der Lebewesen zu. Mit 43 Milliarden Basenpaaren hat der Lungenfisch das bislang größte bekannte tierische und mit 150 Milliarden Basenpaaren die Einbeere Paris japonica das größte bekannte pflanzliche Genom. Die Amöbe Polychaos dubium hat mit 670 Milliarden Basenpaaren (Bp) das größte bekannte Genom überhaupt. Vier Millionen Bp: Escherichia coli; 130 Millionen Bp: Arabidopsis thaliana; 3,2 Milliarden Bp: Mensch; 16 Milliarden Bp: Weizen; 43 Milliarden Bp: Lungenfisch.
● ● ● ▼ ● ● ● ● ● ● ●
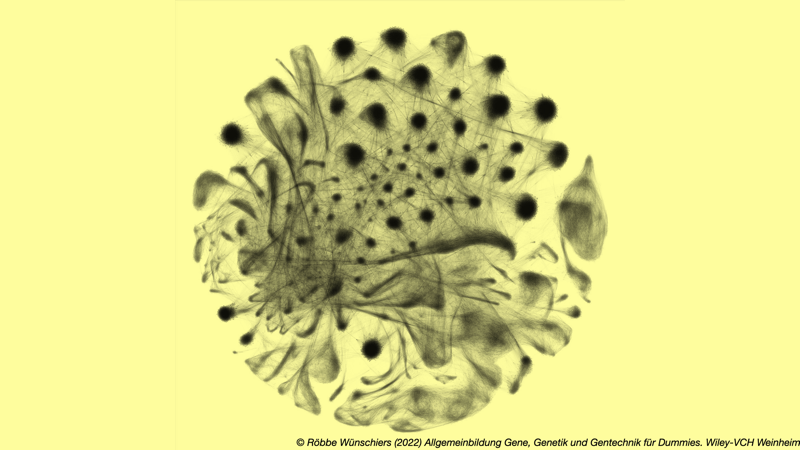
Abb. 3.3: So sieht Komplexität aus. Das Bild gewann im Jahr 2019 den Wellcome Photography Prize. Bild: Carly Ziegler, Alex Shalek, Shaina Carroll vom Massachusetts Institute of Technology und Leslie Kean, Victor Tkachev und Lucrezia Colonna vom Dana-Farber Cancer Institute. Die kreisförmige Anordnung zeigt etwa hunderttausend Zellen des Rhesusaffen. Jede Zelle ist ein Bildpunkt, der erst beim Vergößerung sichtbar werden würde. Mit jeder Zelle sind genetische und phänotypische Merkmale verknüpft. Alle Zellen sind mit einer Linie miteinander verbunden. Je ähnlicher sich zwei Zellen in Hinblick auf ihre Merkmale sind, desto näher werden sie zusammengerückt. So entstehen Anhäufungen ähnlicher Zellen.
● ● ● ● ▼ ● ● ● ● ● ●
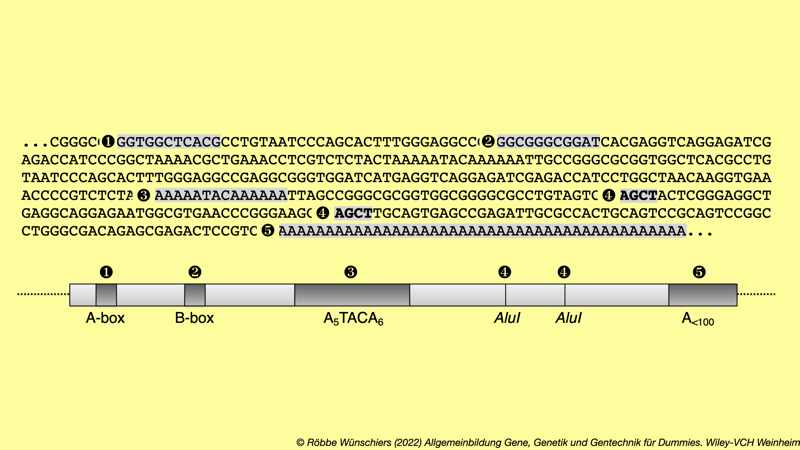
Abb. 3.4: Springende Gene in unserem Genom. Lassen Sie sich von diesem sehr sehr detaillierten Blick in unser Genom nicht abschrecken. Ich möchte Ihnen einfach einmal zeigen, wie ein springendes Gen in »echt« aussieht. Dieses sogenannte Alu-Element ist die häufigste, auf allen Chromosomen über eine Millionen mal wiederholt vorkommende DNA-Sequenz. Zwar gibt es immer kleine Variationen in der Abfolge der Basenpaare, aber der Grundaufbau, hier grau unterlegt, ist immer gleich.
● ● ● ● ● ▼ ● ● ● ● ●
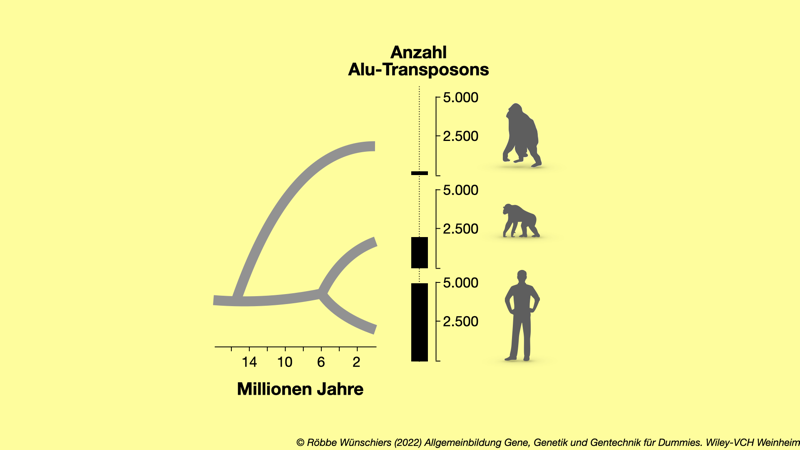
Abb. 3.5: Vergleich der Anzahl der Alu-Elemente, die für Orang-Utans, Schimpansen und Menschen (von oben nach unten) jeweils spezifisch sind.
● ● ● ● ● ● ▼ ● ● ● ●
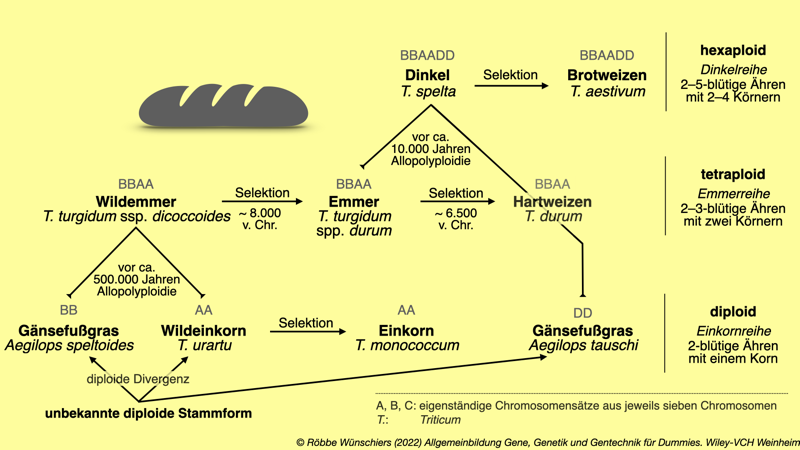
Abb. 3.6: Die Entwicklung der modernen Weizensorten aus ertragsarmen Gänsefußgras und Wildeinkorn. A und B und C kennzeichnen jeweils eigenständige Chromosomensätze, bestehend aus jeweils sieben Chromosomen. Neben der Artbildung durch diploide Divergenz, spielten auch allopolyploide Genomfusionen eine wichtige Rolle bei der Evolution der Genome. T.: Triticum.
● ● ● ● ● ● ● ▼ ● ● ●
Abb. 3.7: Umfang verschiedener Versionen des Humanen Genoms und deren Veröffentlichung. Als im Jahr 2000 die DNA-Sequenz des humanen Erbguts veröffentlicht wurde, was sie noch lange nicht vollständig. Zwanzig Jahre später sind immerhin knapp 96 Prozent komplettiert.
● ● ● ● ● ● ● ● ▼ ● ●
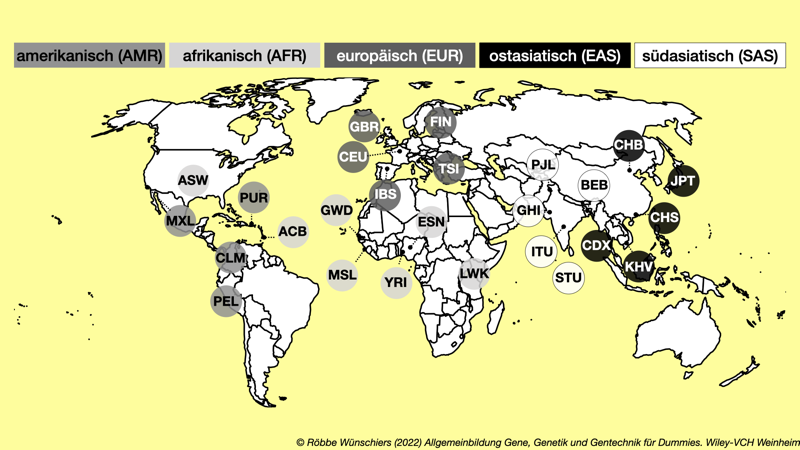
Abb. 3.8: Herkunft der 1.000 Genome. Aus sechsundzwanzig Populationen wurden insgesamt 2.504 Individuen sequenziert. Aus 26 Populationen wurden insgesamt 2.504 Individuen sequenziert. Die Größe der Kreise entspricht der beobachteten Anzahl von Sequenzvarianten, die zwischen 12 und 24 Millionen variierte. Der Drei-Buchstaben-Code bezeichnet die Region. Für Europa (EUR) sind dies: Einwohner von Utah mit nord- und westeuropäischer Abstammung (CEU); Briten in England und Schottland (GBR); Finnen (FIN); Iberische Populationen in Spanien (IBS); Toskana in Italien (TSI).
● ● ● ● ● ● ● ● ● ▼ ●
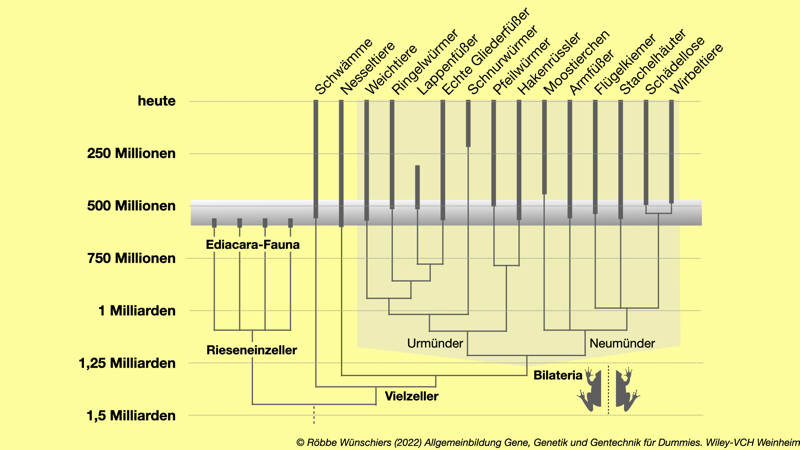
Abb. 3.9: Die kambrische Artenexplosion (Radiation) bei den Tieren. Im Erdzeitalter des Kambriums vor etwa 541 Millionen Jahren kam es zu einer enormen Zunahme von Körperbauplänen. Die Grundlage dafür waren neue genetische Schaltkreise. Besonders erfolgreich war die Radiation der Gruppe der Bilateria. Diese zeichnen sich dadurch aus, das die linke und rechte Hälfte eines Organismus zueinander spiegelsymmetrisch (bilateralsymmetrisch) sind. Die dicken Balken geben Lebenszeiträume der Stämme an, die dünnen Linien deren evolutionäre Verwandtschaft.
● ● ● ● ● ● ● ● ● ● ▼
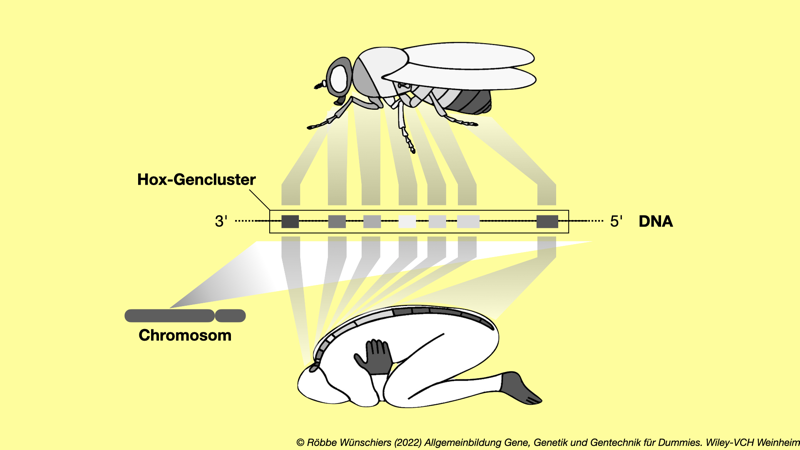
Abb. 3.10: Das Hox-Gencluster. Eine sehr vereinfachte Darstellung der Kolinearität der Hox-Gene. In derselben Reihenfolge, in der die Hox-Gene auf dem Chromosom liegen, werden sind sie auch entlang der Körperachse während der Individualentwicklung aktiv. Die sieben dargestellten Gene stellen den evolutionären Ausgangspunkt dar. Die Taufliege Drosophila hat ein Cluster mit acht Hox-Genen; der Mensch vier Cluster mit insgesamt 39 Hox-Genen.Verändert nach Hueber et al. (2010) doi: 10.1371/journal.pone.0010820. Kapitel 4: Safety First: Gentechnik und Gesetze
▼ ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ●
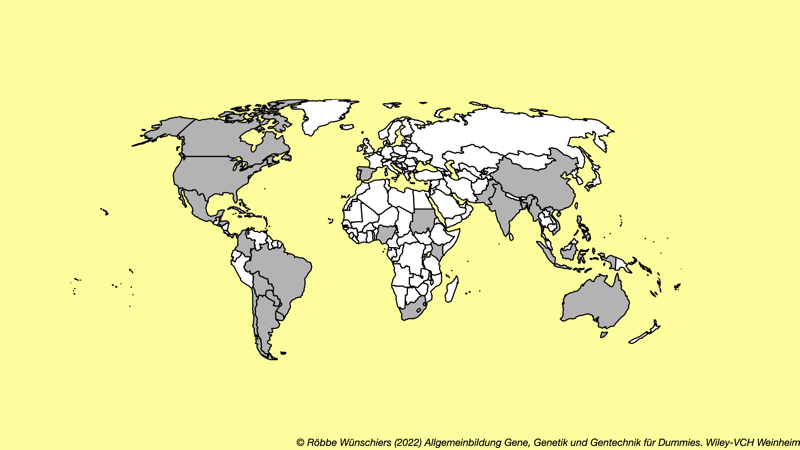
Abb. 4.1: Anbau gentechnisch veränderter Nutzpflanzen weltweit. In den grau gekennzeichneten Staaten werden rund 22 verschiedene Pflanzenarten angebaut, allen voran Soja (50%), Mais (31%) und Baumwolle (13%). Von den insgesamt knapp 190 Millionen Hektar weltweit entfallen vierzig Prozent auf die USA (überwiegend Mais und Soja), 26 Prozent auf Brasilien (überwiegend Soja), zwölf Prozent auf Argentinien (überwiegend Soja), sieben Prozent auf Kanada (überwiegend Raps) und sechs Prozent auf Indien (überwiegend Baumwolle). Die Flächen der Europäischen Union in Spanien und Portugal machen weniger als ein Prozent aus und beschränken sich auf den Anbau des Bt-Mais MON810. Quelle: ISAAA.org.
● ● ▼ ● ●
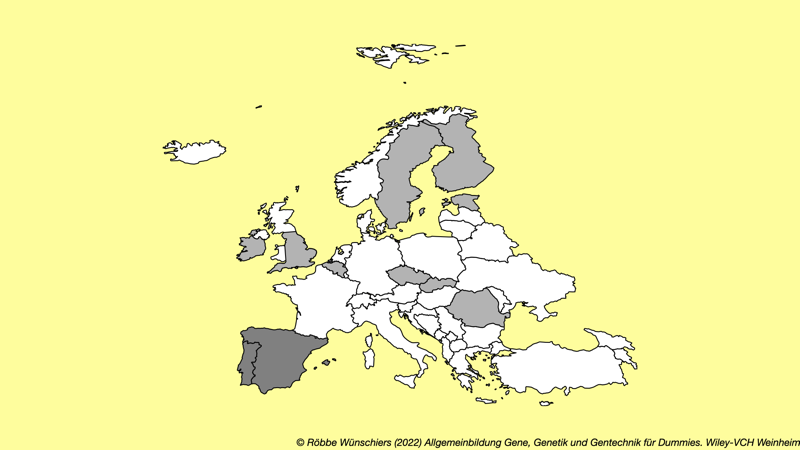
Abb. 4.2: Länder der Europäischen Union in denen der Anbau gentechnisch veränderter Pflanzen erlaubt ist (hellgrau) beziehungsweise in denen sie aktuell angebaut werden: Spanien und Portugal. In beiden Ländern nimmt der Anbau seit 2016 kontinuierlich ab und liegt aktuell bei rund hunderttausend Hektar Bt-Mais MON810 – andere Pflanzen werden nicht angebaut. In allen anderen Ländern ist der Anbau verboten. Quelle: ISAAA.org.
● ● ● ▼ ●
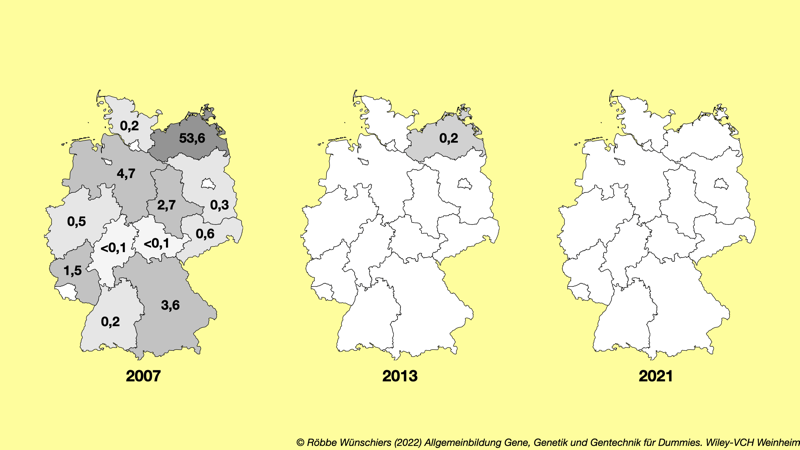
Abb. 4.3: Anbau oder experimentelle Freisetzung gentechnisch veränderter Pflanzen in Hektar in Deutschland. Das Standortregister wird vom Bundesamt für Verbraucherschutz und Lebensmittelsicherheit seit 2005 geführt. Von 2005 bis 2008 wurde der Bt-Mais MON810 von Monsanto (heute Bayer) und von 2010 bis 2011 die Amylopektinkartoffel Amflora von der BASF kommerziell angebaut. Seit 2014 gibt es in Deutschland weder Anbau noch experimentelle Freisetzungen.
● ● ● ● ▼
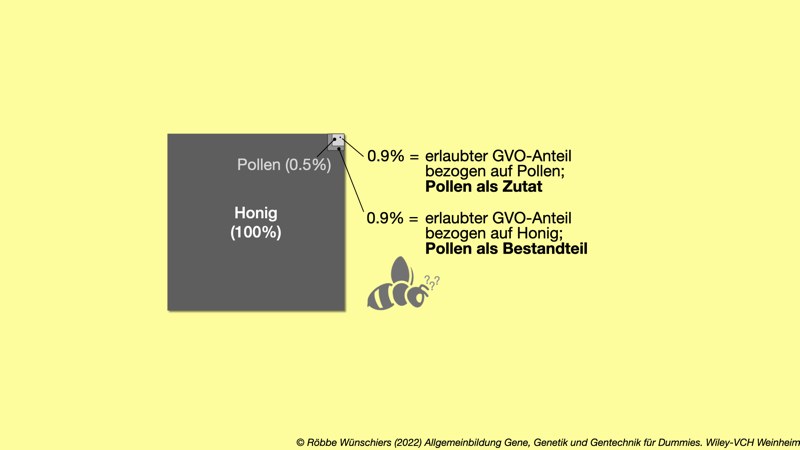
Abb. 4.4: Die 0,9 Prozent Regel bei Pollen als Zutat oder Bestandteil von Honig. Die EU-Verordnung lässt 0,9 Prozent als unvermeidliche »Verunreinigung« einer Zutat mit gentechnisch veränderten Organismen (GVO) zu. Wenn der GVO in der EU nicht zugelassen ist, gilt allerdings die Nulltoleranz. Von 2011 bis 2014 galt Pollen im Honig als Zutat. Kapitel 5: Drei Gene verändern die Welt
▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Abb. 5.1: Restriktionsenzyme erkennen spiegelbildliche oder revers invertierte Palindrome. Sie schneiden die DNA-Doppelhelix, wobei entweder glatte (englisch: blunt) oder klebrige (englisch: sticky) Enden verbleiben. HindII war das erste isolierte und charakterisierte Restriktionsenzym. Es wurde aus dem Bakterium Haemophilus influenzae isoliert. EcoRI stammt aus dem Escherichia coli Stamm RY13 und BbuB31I aus Borrelia burgdorferi.
● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ●
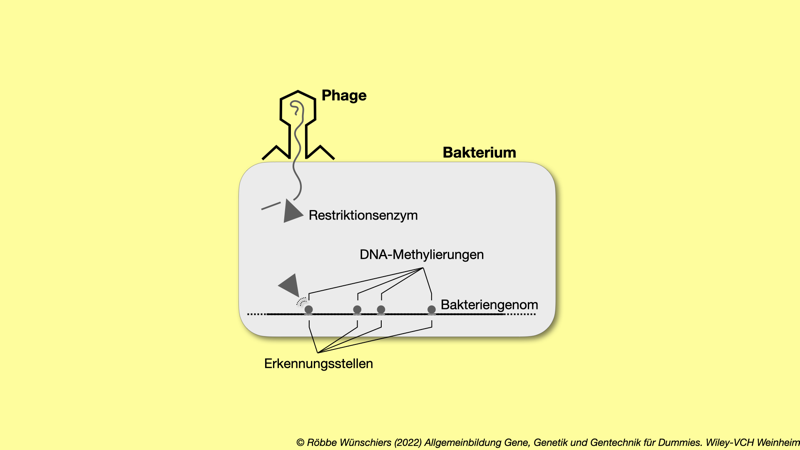
Abb. 5.2: Restriktionsenzyme schneiden eindringende Phagen-DNA an den Erkennungssequenzen (englisch: restriction sites). Das eigene Genom ist durch Methylierungen der Erkennungsstellen geschützt. Darüberhinaus kommen die Erkennungssequenzen im Bakteriengenom nur selten vor, sie sind unterrepräsentiert.
● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ●
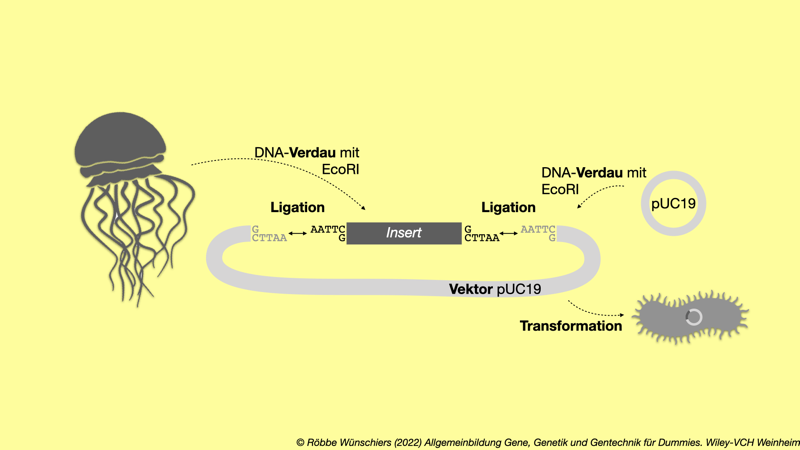
Abb. 5.3: Trans-Gentechnik: Ein DNA-Anschnitt aus der Qualle wird in das Plasmid pUC19 legiert und anschließend ein Bakterium mit dem Plasmid transformiert. Werden bakterielle Vektoren, wie beispielsweise das Plasmid pUC19, aus Bakterien isoliert und mit demselben Restriktionsenzym (hier EcoRI) geschnitten (wir sagen: verdaut) wie die DNA aus einem Quellorganismus, dann ermöglichen insbesondere die klebrige Enden die Ligation dieser DNA-Fragmente (englisch: inserts) in den Vektore. Ligasen sind Enzyme, welche die DNA-Stränge chemisch miteinander verbinden. Der jetzt rekombinante Vektor kann wieder von einem Bakterium aufgenommen werden. Dieser Prozess wird als Transformation bezeichnet.
● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ●
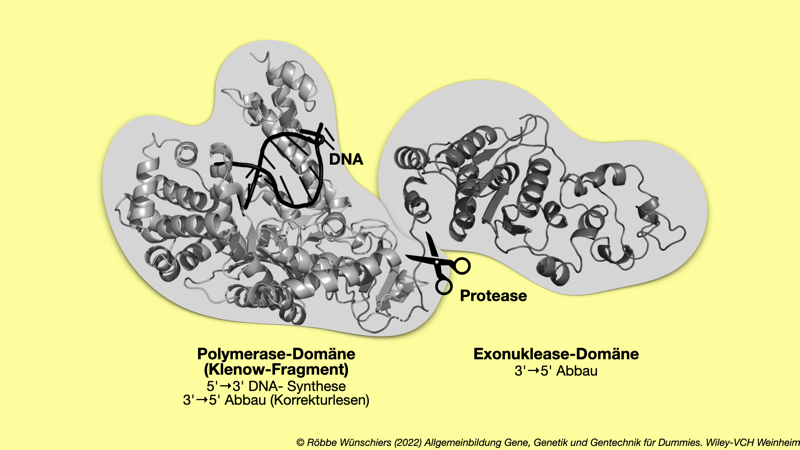
Abb. 5.4: Submolekulare Struktur der DNA-Polymerase I aus dem Bakterium Thermus aquaticus. Man kann gut die beiden Proteindomänen erkennen, die mit ein paar Aminosäuren miteinander verbunden sind. Nach Behandlung des Enzyms mit der Protease Subtilisin, wird die Exonnuklease-Domäne abgetrennt und kann so von der biotechnologisch wichtigen Polymerase-Domäne abgetrennt werden. Subtilisin wird übrigens auch heute noch unter anderem in Waschmitteln eingesetzt.
● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ●
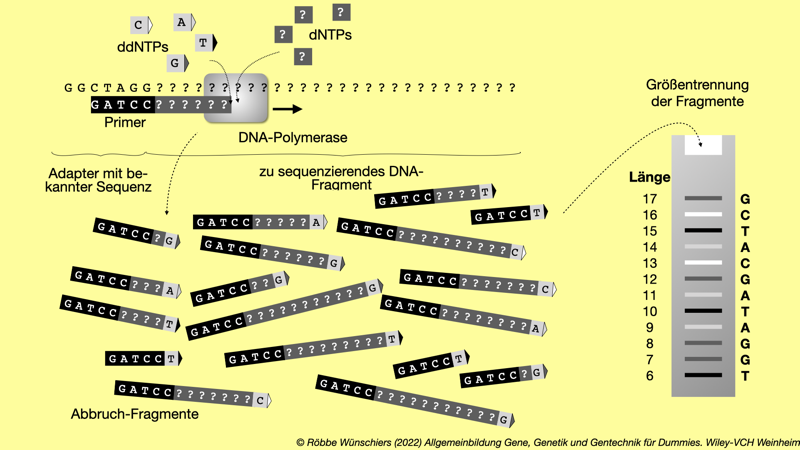
Abb. 5.5: Sanger-Sequenzierung. Der Klassiker der DNA-Sequenzierungsmethoden und noch immer weit verbreitet, um »eben mal was zu sequenzieren«. Ausgehend von einem etwa 25 Nukleotide langen Primer synthetisiert die DNA-Polymerase einen DNA-Strang. Wird statt den normalen dNTPs ein ddNTP eingebaut, bricht die Synthese ab und ein neuer Zyklus beginnt. Die ddNTPs sind mit unterschiedlichen Fluoreszenzfarbstoffen markiert. So ist erkennbar, bei welchem Nukleotid die DNA-Polymerase die Synthese abgebrochen hat. Werden die synthetisierten Fragmente der Länge nach sortiert, kann anhand der Fluoreszenzfarben die Sequenz ermittelt werden. Das geschieht in der Regel in Kapillaren und mit Lasern und lässt sich prima automatisieren. In meinem Beispiel sind die Sequenzen der Anschaulichkeit halber sehr kurz.
● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ●
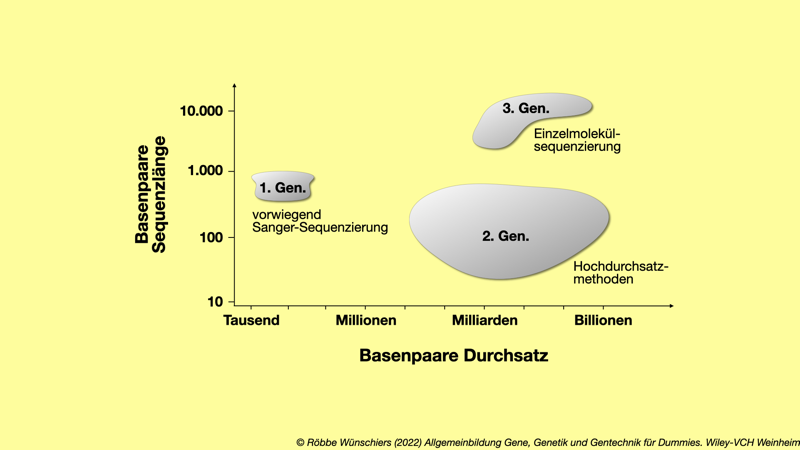
Abb. 5.6: Drei Generationen DNA-Sequenzierung. Technologiesprünge führten zu immer längeren kontinuierlich gelesen DNA-Abschnitten (englisch: read length) und immer mehr erzeugten Sequenzen pro Tag.
● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ●
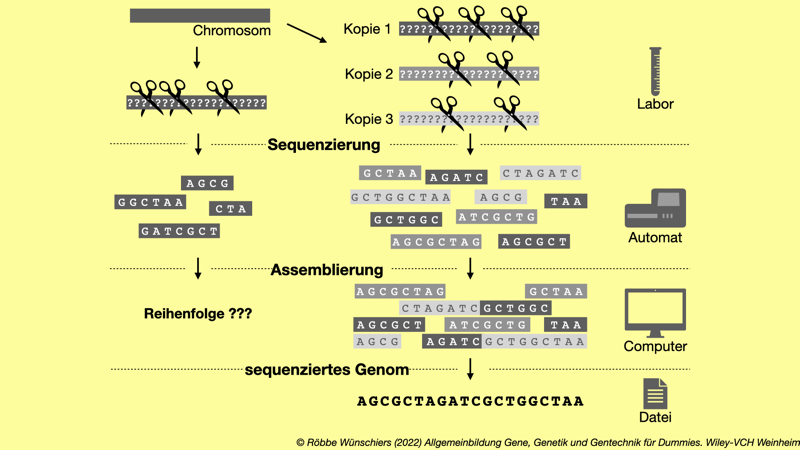
Abb. 5.7: Das Sequenzierpuzzle. Ein Genom kann nicht durchgehend von Anfang bis Ende gelesen werden. Vor der Sequenzierung wird es fragmentiert und die Fragmente sequenziert. Im Gegensatz zur Resequenzierung liegt bei der hier gezeigten de-novo Sequenzierung kein Referenzgenom, also keine Vorlage, vor. An überlappenden Stellen werden die Reads zu einer Gesamtsequenz zusammengesetzt. Für die Genomsequenzierung braucht es Laborarbeit, Geräte-Hightech und Bioinformatik.
● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ●
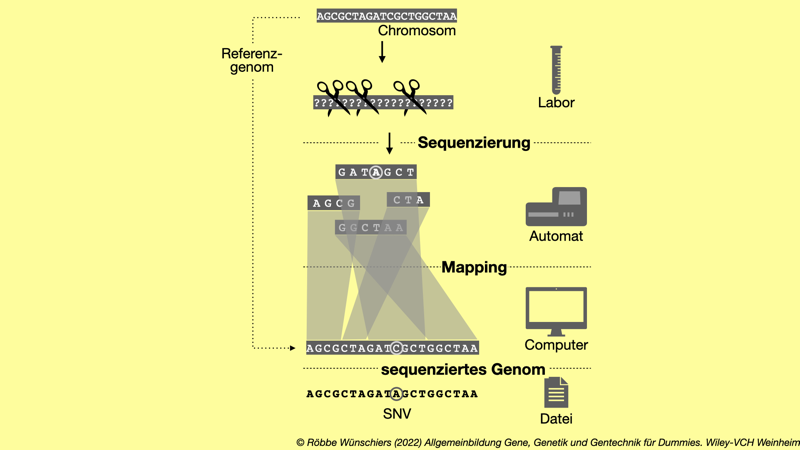
Abb. 5.8: Resequenzierung. Liegt bereits ein Referenzgenom vor, dann können die während der Sequenzerung erhaltenden Reads recht einfach auf die bekannte Sequenz des Referenzgenoms gemappt werden. Das geht auch, wenn kleine Abweichungen von der Referenzsequenz, zum Beispiel SNPs oder allgemein Einzelnukleotidvariationen (SNV; englisch: single nucleotide variance), vorliegen.
● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ●
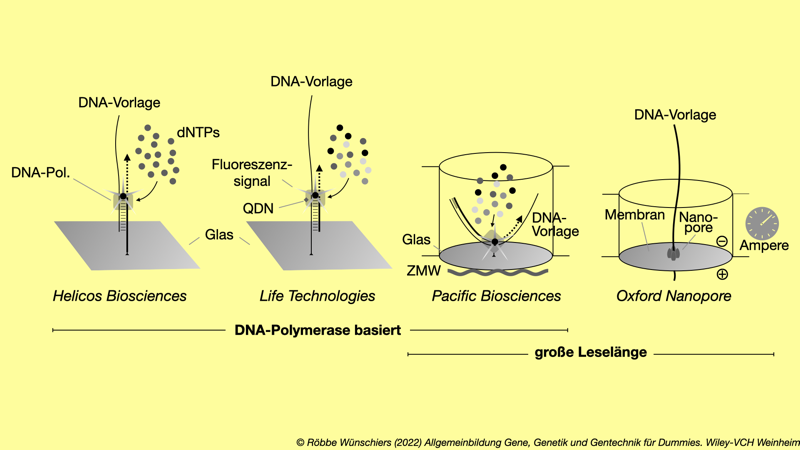
Abb. 5.9: Einzelmolekülsequenzierung. Bei den Sequenziermethoden der dritten Generation werden einzelne DNA-Moleküle (Vorlage) sequenziert. Es entfällt eine vorhergehende Vervielfältigung, die Fehler in die Datenanalyse einführen kann. Drei Firmen arbeiten mit immobilisierten, an eine Glasoberfläche gebundenen DNA-Polymerasen. Hier erfolgt die Erkennung der DNA-Sequenz über die optische Messung des eingebauten Nukleotids (dNTP). Bei Oxford Nanopore dagegen wir die DNA durch Nanoporen gezogen und der sequenzabhängig geänderte Stromfluss gemessen. QDN = Quantum Dots Nanocrystals; ZMW = Zero Mode Waveguide.
● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ●
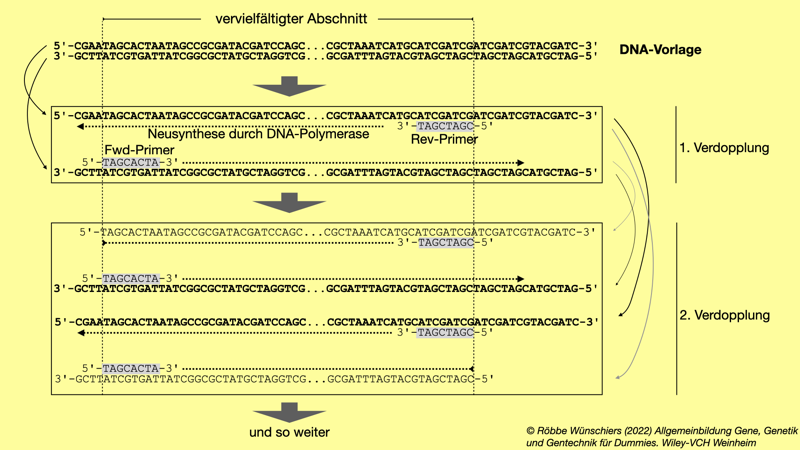
Abb. 5.10: Bei der Polymerasekettenreaktion (PCR) wird ein von den Primern eingeschlossener Bereich der DNA-Vorlage vervielfältigt. Nach zwanzig Zyklen liegen theoretisch 220 = 1.048.576 DNA-Doppelhelices vor. Die Primer sind hier verkürzt dargestellt; im Experiment sind sie um die 25 Nukleotide lang.
● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ●
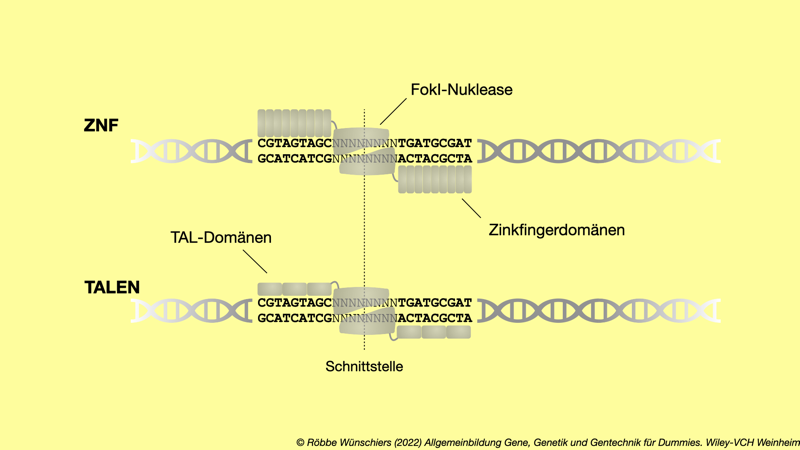
Abb. 5.11: Zinkfingernukleasen (ZNF) und Transcription Activator-Like Effector Nucleases (TALENs) waren die ersten Genscheren, bevor das CRISPR/Cas-System entdeckt und anwendbar gemacht wurde. Ihr Nachteil: Die DNA-Bindedomänen bestehen aus Protein, was das Design und die Herstellung erheblich erschwert.
● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ●
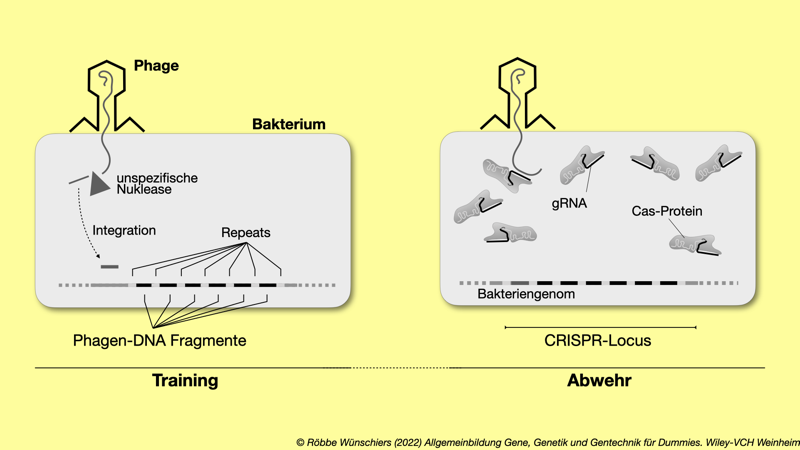
Abb. 5.12: Das CRISPR/Cas-Immunsystem der Bakterien. Wird ein Bakterium von einem ihm unbekannten Phagen befallen, dann wird die Phagen-DNA wird unspezifisch durch Restriktionsenzyme zerschnitten. Dadurch entstandene DNA-Fragmente können in den CRISPR-Locus des Bakteriengenoms eingebaut werden. Wird das Bakterium, oder ein Nachfahre, von dem gleichen Phagen erneut infiziert, können die mit guideRNA (gRNA) beladenen Cas-Nukleasen (Genscheren) die Phagen-DNA zerschneiden. Die Spezifität wird durch die gebundenen gRNA-Moleküle erreicht.
● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ●
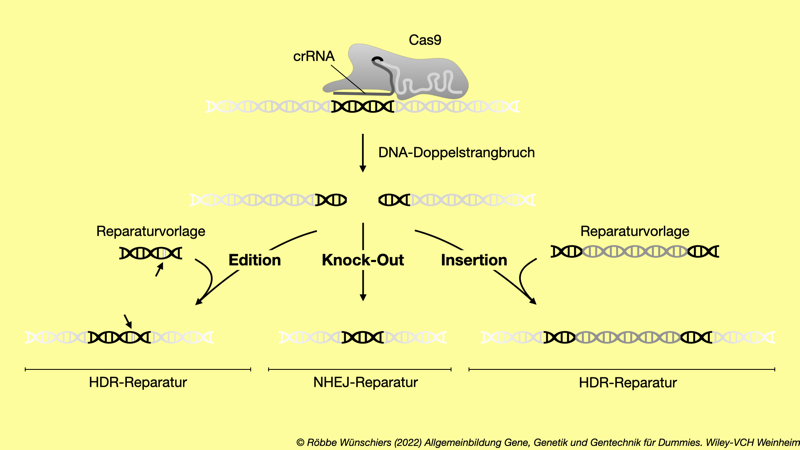
Abb. 5.13: Arbeiten mit der Genschere CRISPR/Cas. Die crRNA arbeitet wie eine Sonde und leitet die Genschere, das Cas9-Protein, an die Zielsequenz auf der doppelsträngigen DNA. Nachdem die DNA geschnitten wurde, wird sie entweder durch den HDR-Mechanismus (englisch: homology directed repair) oder den NHEJ-Mechanismus (englisch: non-homologous end joining) wieder zusammengesetzt. Diese Reparatur kann durch zugesetzte Reparaturvorlagen beeinflusst werden. NHEJ führt zu einer ungenauen Veränderung der Gensequenz und damit in der Regel zum Abschalten des Gens (Knock-out) durch Mutationen. Über den HDR-Mechanismus können Basenpaare gezielt editiert oder ganze Gene inseriert werden.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ●
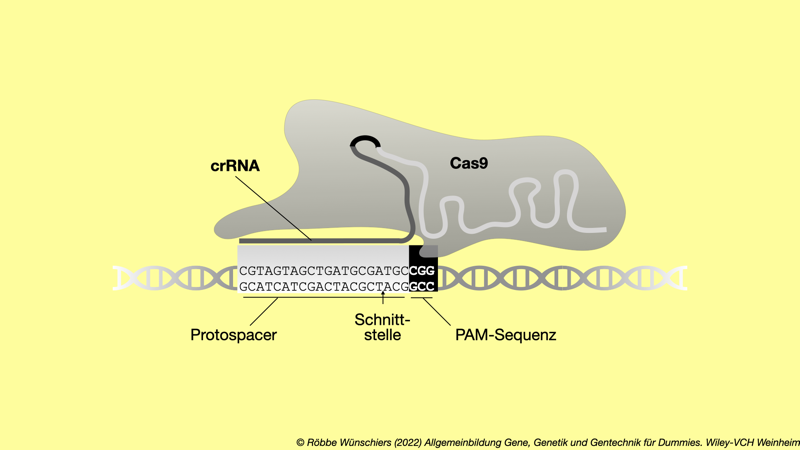
Abb. 5.14: Drei Basenpaare entscheiden über Leben und Tod. Neben der Zielsequenz, dem Protospacer, die von der crRNA erkannt wird, liegt die PAM-Sequenz. Sie wird von dem Cas-Protein erkannt.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼
Abb. 5.15: Der Kampf von Cas9 gegen die Phagen erinnert an Hamlet, Prinz von Dänemark von William Shakespeare. »Sich waffnend gegen eine See von Plagen«? Es ist nur eine kleine Buchstabenmutation, die uns zum Phagen bringt. Kapitel 6: Bändigung des Zufalls
▼
Kunst am Buch – von Kerstin Zentner. Kapitel 7: Dolly war kein Clown
▼ ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ●
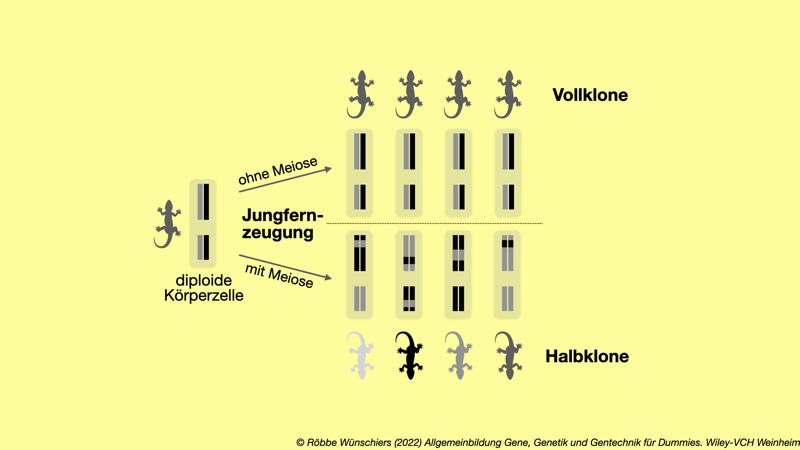
Abb. 7.1: Vollklone sind exakte genetische Kopien ihrer Mutter. Halbklone tragen dagegen nur die Hälfte der Allele der Mutter, diese aber dafür doppelt.
● ● ▼ ●
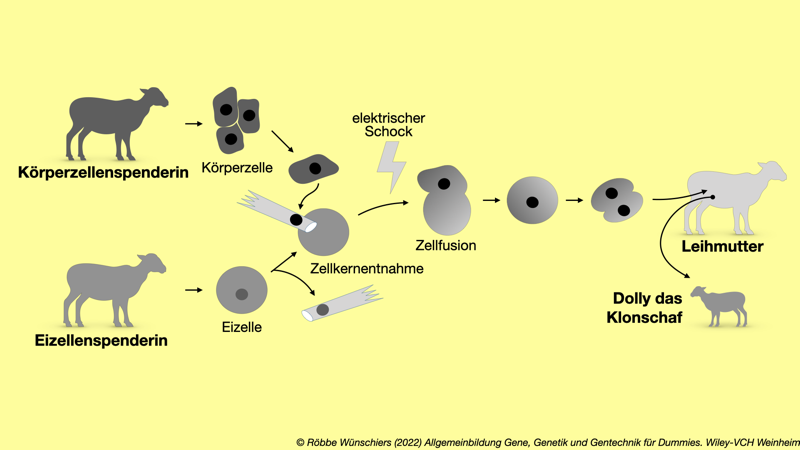
Abb. 7.2: Das Klonen von Dolly, dem Schaf. Beim Klonen von Tieren werden genetisch identische Kopien erstellt. Dazu wird aus einer Spendereizelle der Zellkern samt Erbgut entfernt. Aus der Körperzelle des zu klonierenden Tieres wird der Zellkern in die entkernte Eizelle eingefügt – das Verfahren heißt somatischer Zellkerntransfer. Mittels eines elektrischen Stromstoßes wird die Hybridzelle zur Zellteilung angeregt. Der Zellhaufen wird einer Leihmutter implantiert, die dann den Klon gebärt.
● ● ● ▼
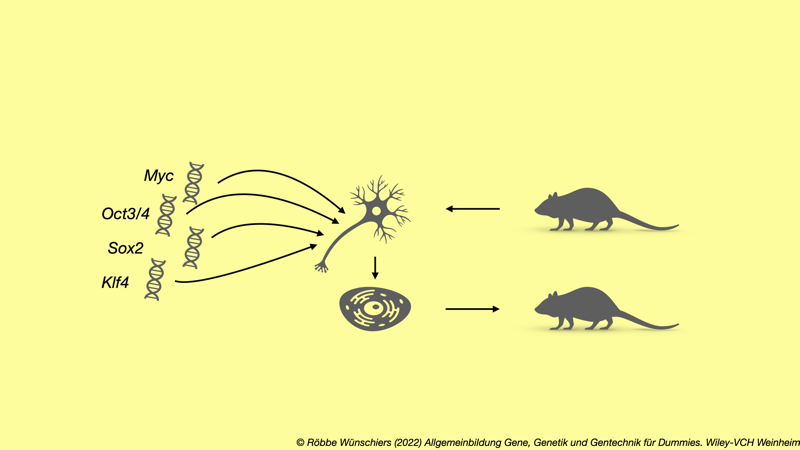
Abb. 7.3: Induzierte pluripotente Stammzellen (iPS). Das Einbringen von vier Genen mittels retroviralen Vektoren in ausdifferenzierte Körperzellen programmiert diese zu pluripotenten Stammzellen um. Aus diesen kann wieder ein kompletter Organismus regeneriert werden. Kapitel 8: Unsere DNA als Glaskugel
▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ●
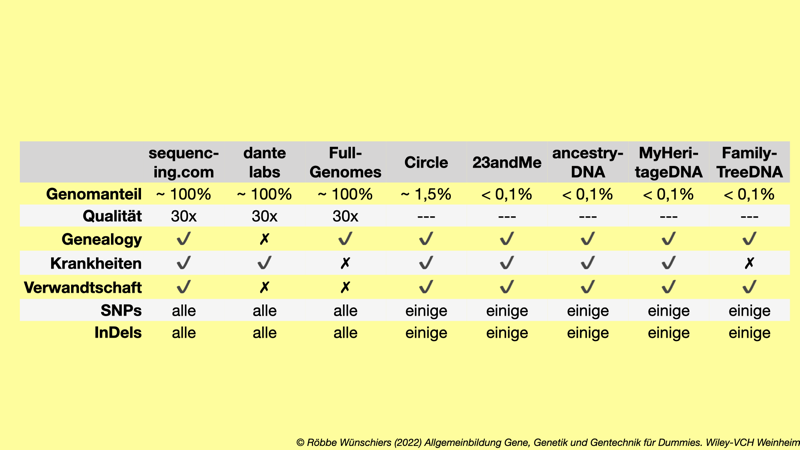
Tab. 8.1: Ausgewählte Dienstleister, die DNA-Analysen für Privatpersonen anbieten. Nur wenige Firmen bieten die Sequenzierung und Analyse des gesamten Erbguts an. Die Kosten schwanken zwischen 89 und 599 Euro. Am Black Friday ist es natürlich billiger.
● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ●
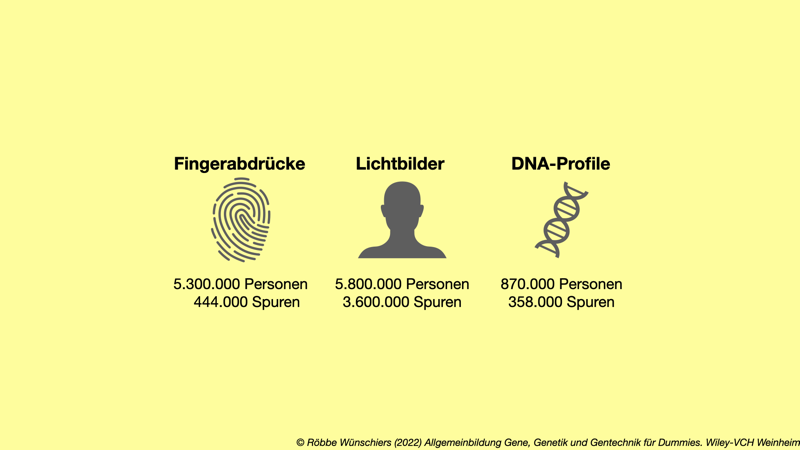
Abb. 8.1: Erkennungsdienstliche Datenbanken des Bundeskriminalamts speichern behördlich erhobene Fingerabdrücke, Lichtbilder und DNA-Profile von Personen sowie Daten von Tatortspuren. Stand März 2020.
● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ●
Abb. 8.2: Der Genetiker Diethard Tautz gratuliert dem Autor zur Habilitation in Genetik an der Universität zu Köln im Jahr 2006. Zuvor gab es nach meiner Prüfungsvorlesung zur Epigenetik eine aufreibende Diskussion zum Lamarckismus. Tautz ist jetzt Direktor am Max-Planck-Institut für Evolutionsbiologie in Plön.
● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ●
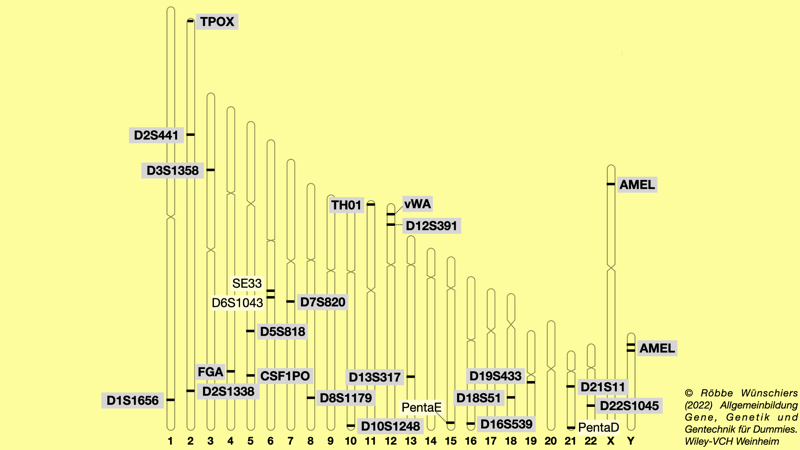
Abb. 8.3: Bezeichnung der Genorte (Loci), die für den genetischen Fingerabdruck verwendet werden. Seit 2017 kommen zwanzig verschiedene STRs (short tandem repeats) zum Einsatz, die als CODIS (Combined DNA Index System) bezeichnet werden. Hinzukommt das Amelogeningen (AMEL), dass der Geschlechtsbestimmung dient.
● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ●
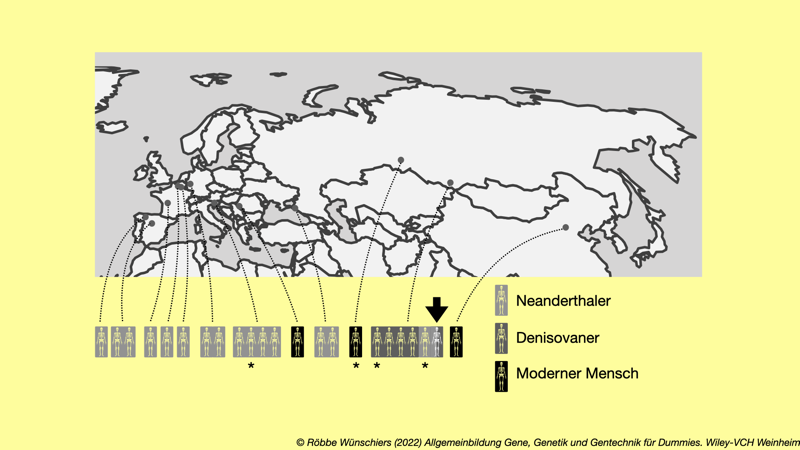
Abb. 8.4: Fundorte unserer Vorfahren. Es sind nur Funde gezeigt, von denen ein Teil des Erbgutes untersucht werden konnte. Mit einem Stern markierten Funde wurden vollständig sequenziert. Mit einem Pfeil markiert ist der Fund eines Mädchens, deren Vater Densisovaner und die Mutter eine Neanderthalerin war.
● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ●
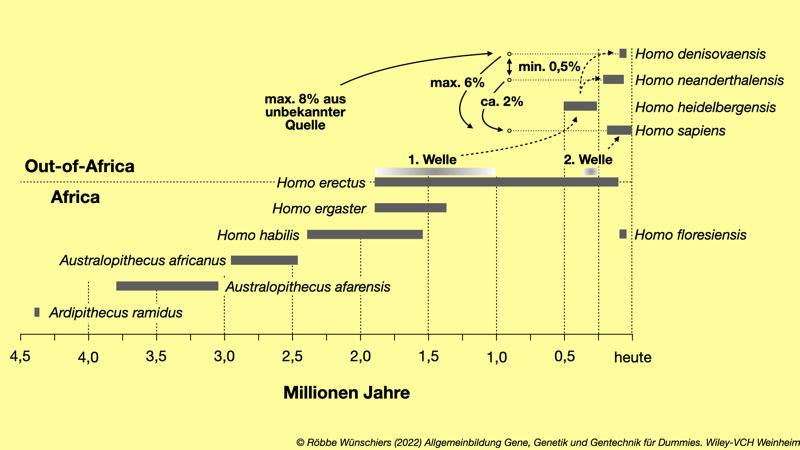
Abb. 8.5: Zeitliches Auftreten der Vorläufer des modernen Menschen (Homo sapiens). Ardipithecus ramidus gilt als letzter gemeinsamer Vorfahr der Menschenaffen: Gorillas, Menschen, Orang-Utans, Schimpansen. Zu Australopithecus afarensis (Südaffe aus Afar) gehört das berühmte Skelett namens »Lucy«. Markiert sind zwei Auswanderungswellen des Homo erectus aus Afrika und die resultierenden Arten. In den letzten fünfzigtausend Jahren lassen sich genetische Flüsse zwischen den Arten nachweisen.
● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ●
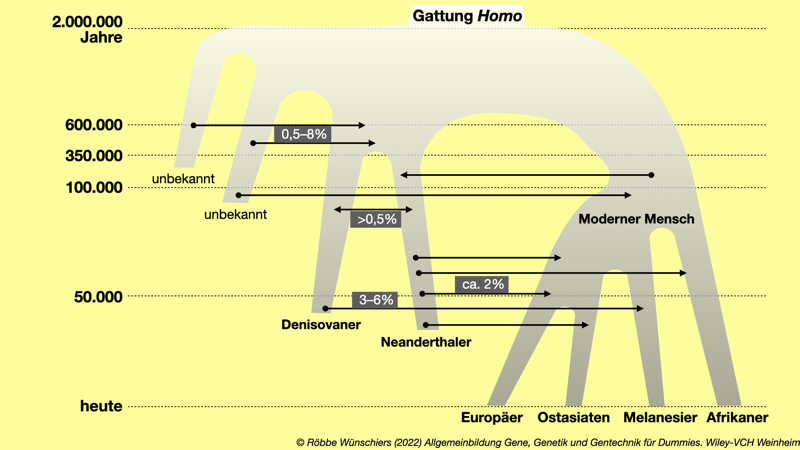
Abb. 8.6: Das Netzwerk der Gattung Homo. Obwohl die ältesten Erbgutinformationen von Vertretern unserer Vorfahren knapp fünfzigtausend Jahre alt sind, können davor stattgefundene Genaustausche durch Berechnungen vorhergesagt werden. Die aktuell lebenden Menschengruppen teilen sich gemeinsame Vorfahren und tauschen selbstverständlich auch Gene untereinander aus. Es gibt auch Hinweise auf Kreuzungen zwischen Neanderthalern und Denisovanern sowie zwischen Denisovanern und einem unbekannten, ausgestorbenen Menschentyp. Melanesien ist ein Kulturraum nördlich von Australien und umfasst unter anderem die pazifischen Inselgruppen Papua-Neuguinea, Fidschi und die Salomonen. Nach Gibbons (2020) doi: 10.1126/science.abb3777
● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ●
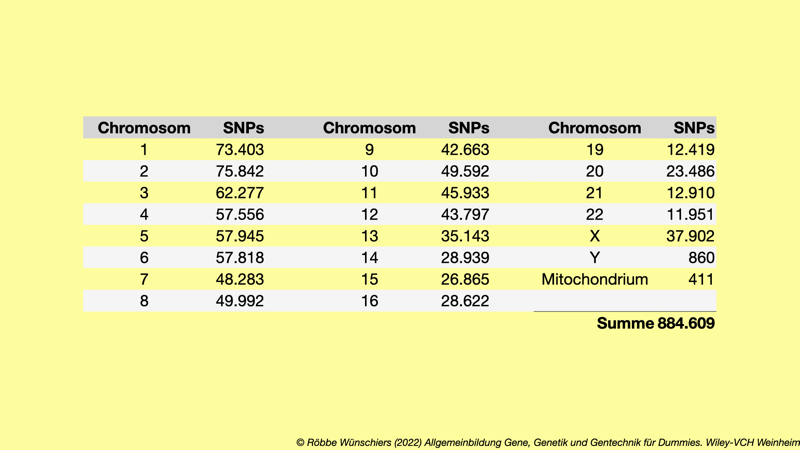
Tab. 8.2: SNPs auf dem DNA-Mikroarray GeneChip 6.0 der Firma Affymetrix. Die Chromosomen sind in gewohnter Weise benannt. »Mt« bezeichnet das Mitochondriengenom.
● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ●
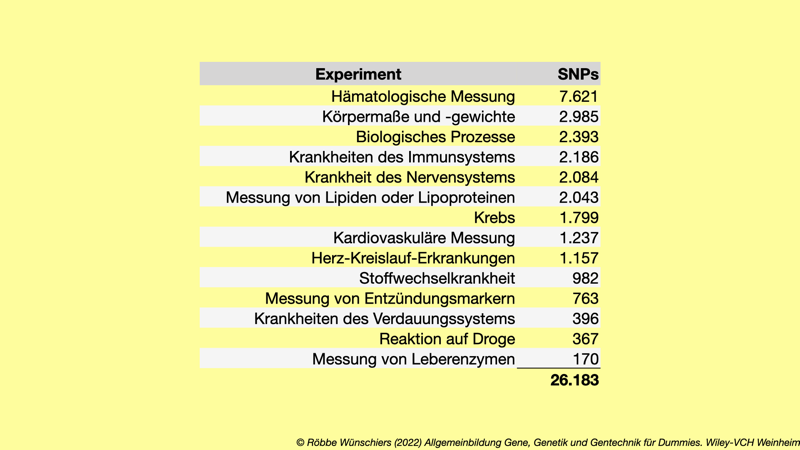
Tab. 8.3: Hoch signifikante SNP-Merkmal-Assoziationen im GWAS-Katalog.
● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ●
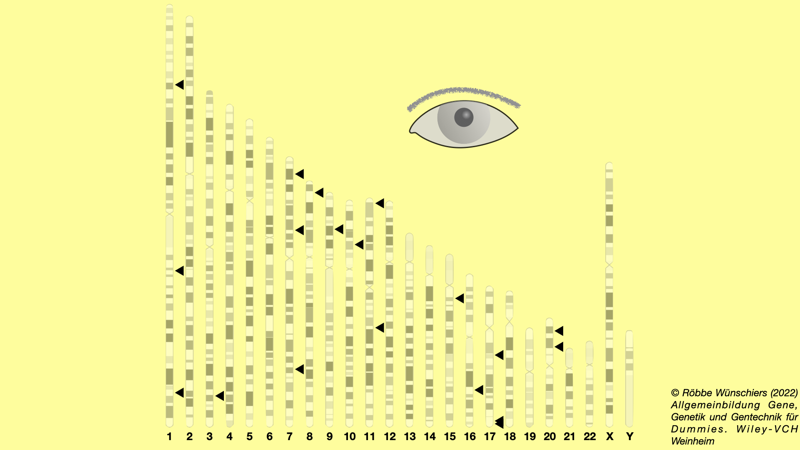
Abb. 8.7: Chromosomenorte, die mit dem Grauen Star in Verbindung stehen. In der Studie wurden 1.754 Genome des UK Biobank Project analysiert. Bei 456 Teilnehmern der Studie wurde der Graue Star operativ behandelt.
● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ●
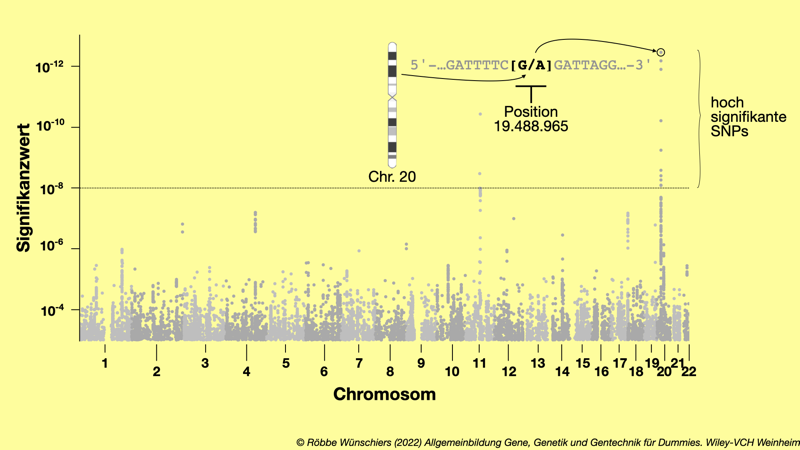
Abb. 8.8: Einzelnukleotidpolymorphismen (SNPs), die mit dem Grauen Star in Verbindung stehen. Dargestellt sind die Zugehörigkeit zu den Chromosomen und der Signifikanzwert (p-Wert) für 12.253 SNPs. Zwei SNPs auf Chromosom 11 und zehn SNPs auf Chromosom 20 stehen hoch signifikant mit dem Auftreten des Grauen Stars in Verbindung. Für diese Studie wurden 11.986 erkrankte und 115.617 gesunde Personen untersucht. Die höchste Signifikanz zeigt der SNP rs4814862 an Position 19.488.965 auf Chromosom 20, der einen G/A-Polymorphismus aufweist. Im Vergleich zur den Ergebnissen der Studie in Abb. 8.7, sind wegen des größeren Studienumfangs die Ergebnisse genauer. Daher sind hier sechs Gene weniger signifikant mit Grauem Star assoziiert.
● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ●
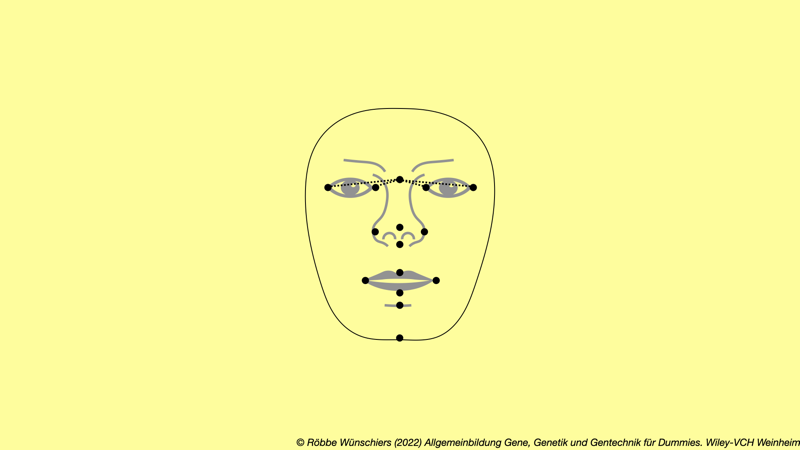
Abb. 8.9: Die dreidimensionalen Abstände markanter Gesichtsmarken charakterisieren ein Gesicht. Die zugrundeliegende Methodik wurde bereits 1879 von dem britischen Naturforscher Francis Galton beschrieben. Besonders die Abstände zwischen den Augenrändern und der Nasenwurzel sind in hohem Maße mit genetischen Varianten korreliert, wie aktuelle Studien zeigen.
● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ●
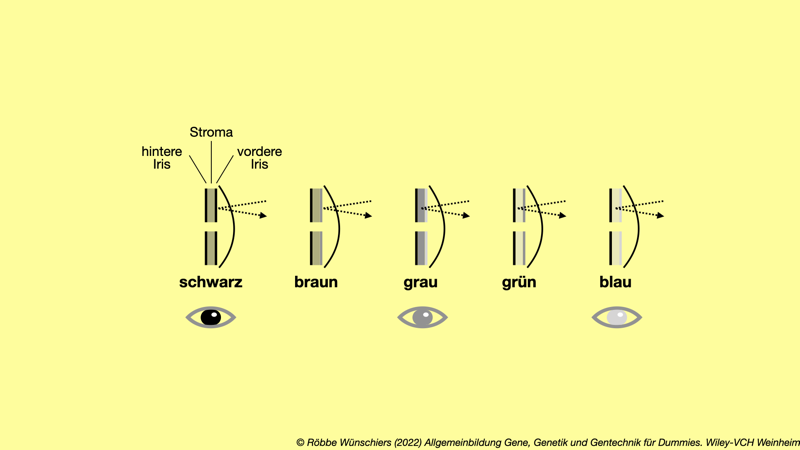
Abb. 8.10: Die Farbe der Augeniris hängt von mehreren Faktoren ab. Je pigmentierter die vordere Iris ist, desto lichtundurchlässigerer ist sie. Tritt Licht durch sie hindurch, bestimmt zusätzlich die Struktur des Stromas die Farbe des reflektierten Lichts. Dieses Gewebe zwischen den beiden Iriden streut das Licht in der gleichen Weise wie Staubpartikel in der Atmosphäre. Blaue Augen und der blaue Himmel haben also denselben Ursprung für ihr Blau.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼
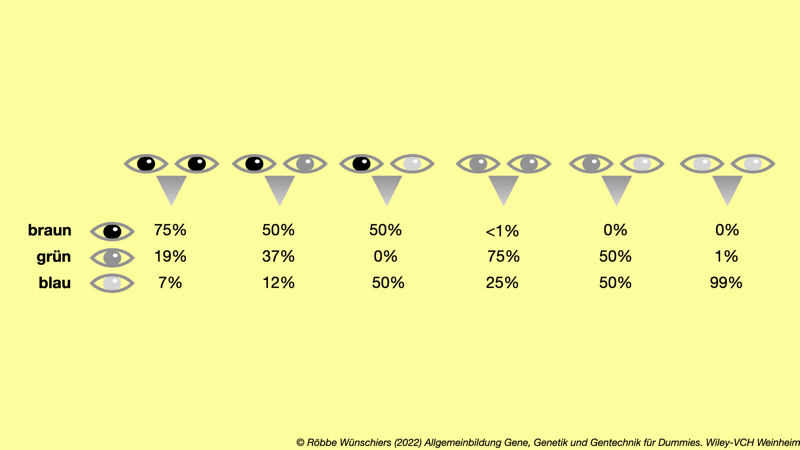
Abb. 8.11: Das klassische und vereinfachte Schema der Vererbung der Augenfarbe. Die beiden Augen in der oberen Zeile symbolisieren jeweils die elterlichen Augen. Darunter sind die üblichen Verteilungen der Irisfarben der Kinder angegeben. Kapitel 9: Künstliche Gene und Organismen
▼ ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ●
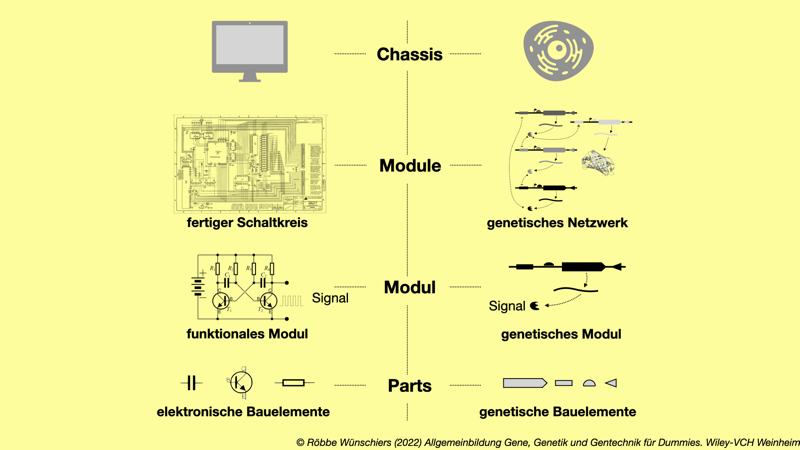
Abb. 9.1: Baukastenprinzip in der synthetischen Biologie. Wie in der Elektronik, sollen standardisierte Bauteile (parts) zu funktionalen Komponenten (Modulen) zusammengesetzt und in Chassis eingebaut werden.
● ● ▼ ● ●
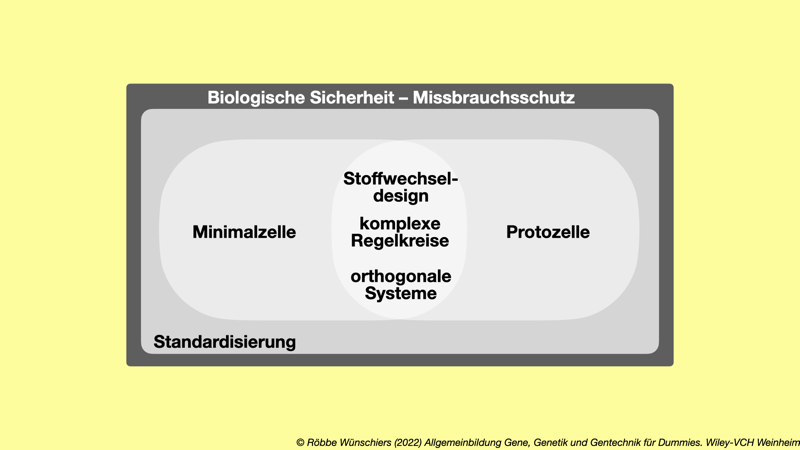
Abb. 9.2: Die sieben Teilgebiete der synthetischen Biologie. Minimalzellen und Protozellen stellen Chassis dar, in denen designte Stoffwechselwege und komplexe Schaltkreise als orthogonale Systeme implementiert werden sollen. Ergänzend ist die Standardisierung der biologischen Bauelemente erwähnt. Alle Entwicklungen werden in Bezug auf ihre biologische Sicherheit und das Missbrauchsrisiko hin kontrolliert.
● ● ● ▼ ●
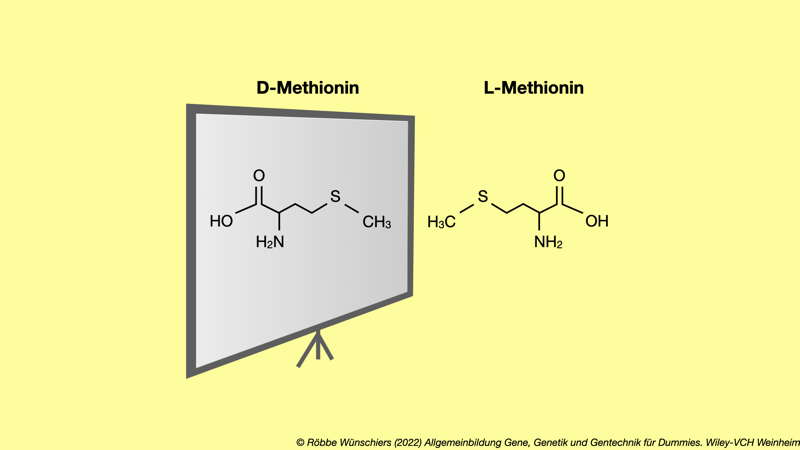
Abb. 9.3: Spiegelbildliche Welten. Alle am Proteinaufbau beteiligten Aminosäuren haben die L-Form. Die dazu spiegelbildliche D-Form existiert zwar in einigen Bakterien als Baumaterial für Zellwände, aber (noch) nicht als Bausteine für Proteine.
● ● ● ● ▼
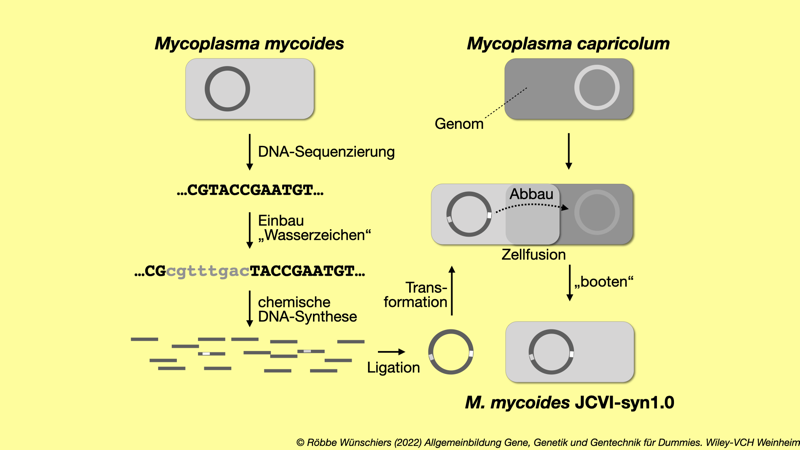
Abb. 9.4: Herstellung von Mycoplasma mycoides JCVI-syn1.0. Nach der vollständigen chemische Synthese des Chromosoms von Mycoplasma mycoides und Rückführung in eine Mycoplasma mycoides Zelle, wurde diese mit Mycoplasma capricolum fusioniert. Neben Wasserzeichen enthält das synthetische Erbgut Code für eine Nuklease, die das M. capricolum Genom abbaut. Mit dem Ablesen (»booten«) des neuen Genom, beginnt daher die genotypische und phänotypische Verwandlung (»Umprogrammierung«) von M. capricolum in M. mycoides Kapitel 10: Künstliche Intelligenz und echte Erkenntnisse
▼ ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ●
Abb. 10.1: Das UK Biobank Project. Rund eine halbe Millionen Briten stellen ihre medizinischen und privaten Daten für eine umfassende Datenanalyse zur Verfügung. Die Datenbank mit den anonymisierten Daten kann von Wissenschaftlern in der ganzen Welt genutzt werden.
● ● ▼
Abb. 10.2: Die Aminosäuresequenz des Cas9-Proteins. Diese 1.372 Aminosäuren lange Proteinkette, die von oben links nach unten rechts verläuft, faltet sich dreidimensional in die Struktur der Cas9-Endonuklease, wie sie auf dem Buchdeckel zu sehen ist. Das Programm AlphaZero kann diese Faltung vorhersagen. Kapitel 11: Gentechnik und Umweltschutz – geht das?
▼ ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ●
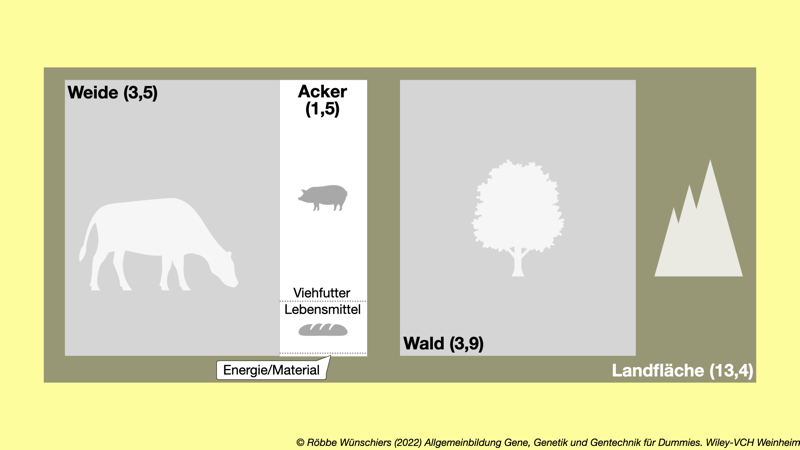
Abb. 11.1: Landnutzung auf unserem Globus. Die Gesamtfläche der Erde beträgt rund 13,4 Milliarden Hektar (134 Millionen km2). Davon entfallen knapp vier Milliarden Hektar auf Wald und knapp fünf Milliarden Hektar auf Agrarflächen. Davon wiederum sind etwa siebzig Prozent Weideland. Auf den 1,5 Milliarden Hektar Ackerfläche werden wiederum zu rund siebzig Prozent Futter- und zu rund achtzehn Prozent Nahrungspflanzen angebaut. Die restlichen zwölf Prozent entfallen auf Energie- und Materialpflanzen. Auf etwa dreizehn Prozent der Ackerfläche werden gentechnisch veränderte Pflanzen angebaut.
● ● ▼ ● ●
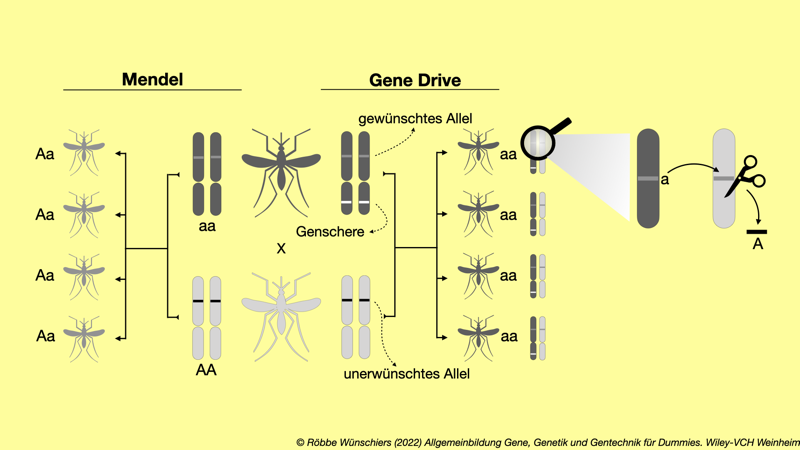
Abb. 11.2: Der Gene Drive umgeht die Mendelsche Vererbung. Statt zu einem Mix mütterlicher und väterlicher Allele (links) führt der Gene Drive bei den Nachkommen dazu, dass das Wildtypallel aus der Population verdrängt wird (rechts). Dies geschieht, indem die Mücke zusätzlich die Gene für die CRISPR/Cas-Genschere im Erbgut trägt.
● ● ● ▼ ●
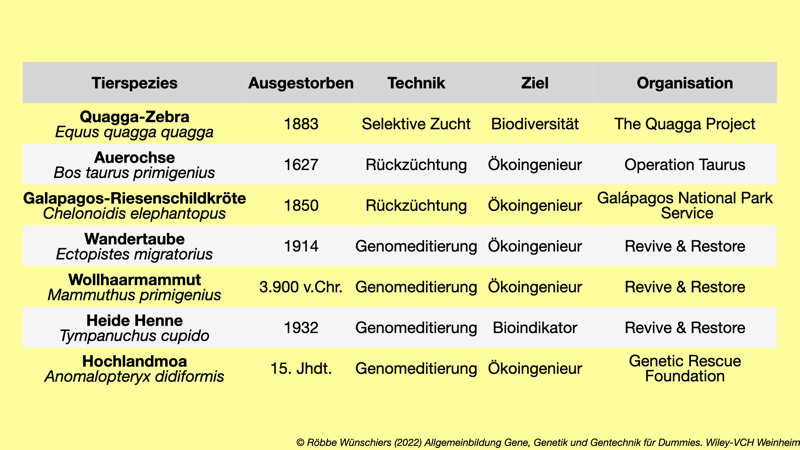
Tab. 11.1: Auswahl aktiver De-Extinction Programme: quaggaproject.org; taurosproject.com; galapagos.org; reviverestore.org; geneticrescue.org Der Erhalt von Ökoingenieuren nutzt gleichzeitig auch anderen Arten in demselben Ökosystem. Sie leisten beispielsweise durch ihr Fraßverhalten einen signifikanten Beitrag zur Ökosystemleistung.
● ● ● ● ▼
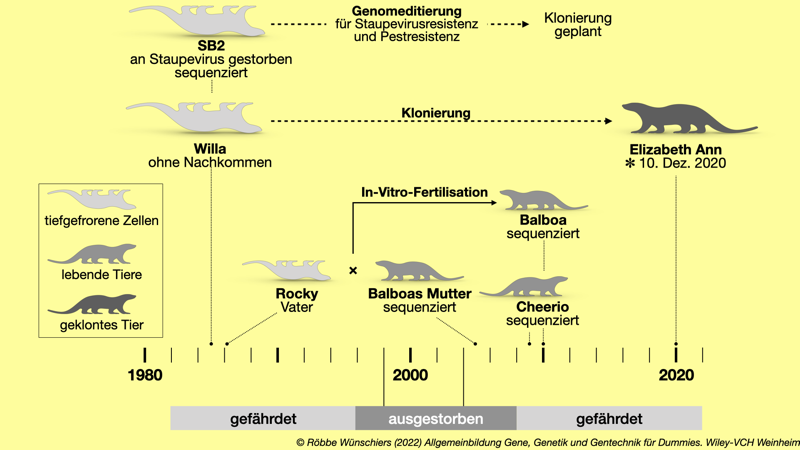
Abb. 11.3: Die Rettung des Schwarzfußiltis. Mitte der 1980er Jahre wurde aus sieben Tieren eine Zuchtpopulation aufgebaut und in den nachfolgenden Jahre ausgewildert. Um die genetische Diversität zu erhöhen, wurde 2010 Balboa über eine künstliche Befruchtung aus tiefgefrorenen Spermien und einer lebenden Mutter gezeugt. Sie wurde zur Weiterzucht verwendet. Im Dezember 2020 gelang die das Klonen eines 1985 gefangenen, verstorbenen und tiefgekühlten Tieres. Kapitel 12: Epigenetik: Darwin und Lamarck vereint
▼ ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ●
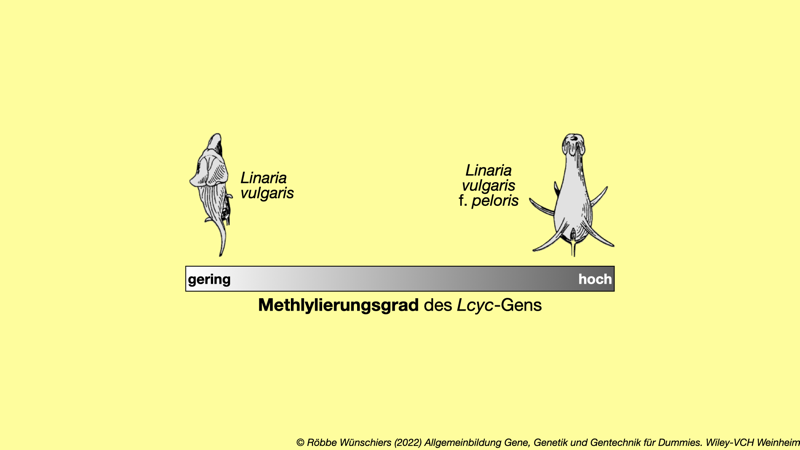
Abb. 12.1: Blütenformen des Echten Leinkrauts (Linaria vulgaris). Zeichnungen von Johann Wolfgang von Goethe (1749–1832). In Abhängigkeit vom Methylierungsgrad des Lcyc-Gens, verwandelt sich die Blütenform von der typischen bilateralsymmetrischen in die von \textsc{Linné} als monsterhaft (peloris) beschriebene radiärsymmetrische Blütenform.
● ● ▼ ● ●
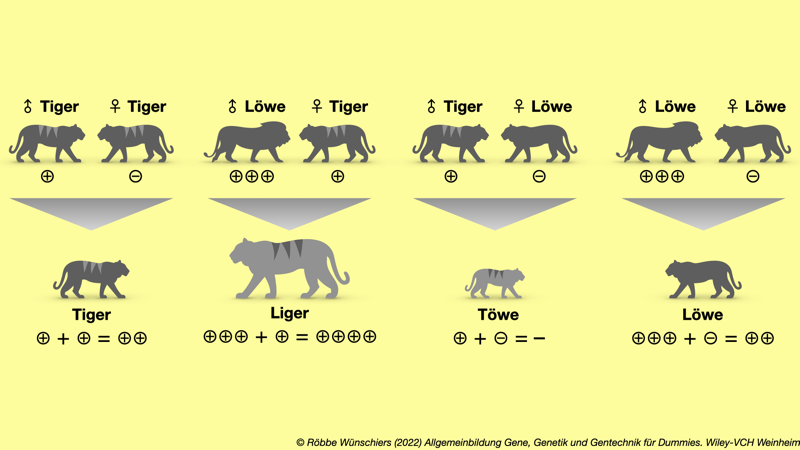
Abb. 12.2: Löwe trifft Tiger. Die Aktivität des Wachstrumsfaktors Igf2 hängt von der Tierart und vom Geschlecht ab. Sie wird über den Grad der Methylierung der DNA reguliert und bestimmt letztlich die Größe der Nachkommen. Liger, eine Kreuzung aus Löwenkater und Tigerkatze, kann bis zu 3,5 Meter lang werden, während Töwen eher kleiner als die Elterntiere sind.
● ● ● ▼ ●
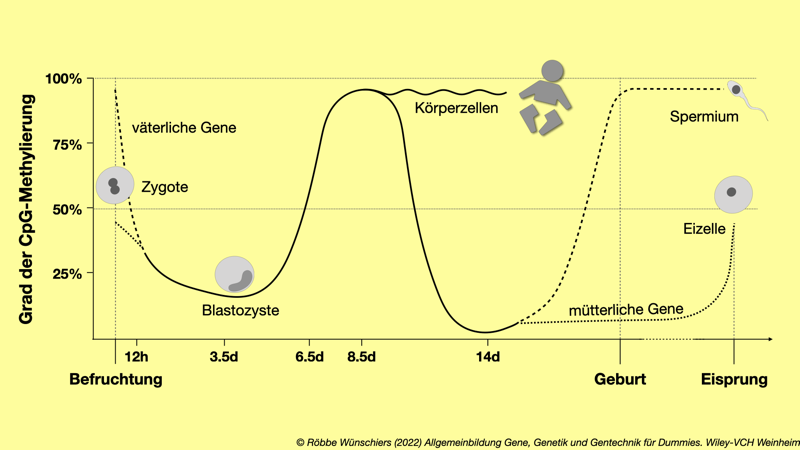
Tab. 12.1: Grad der DNA-Methylierung von der Befruchtung bis zur Geburt. Das Spermiengenom der Maus ist an seinen CpG-Stellen in der DNA bis zu neunzig Prozent methyliert (zwanzig Millionen methylierte Stellen). Nach der Befruchtung wird das väterliche Chromosom innerhalb weniger Stunden durch einen aktiven Prozess vor der DNA-Replikation fast vollständig demethyliert. Im Genom der reifen Eizelle sind etwa vierzig Prozent aller CpG-Stellen methyliert.
● ● ● ● ▼
Abb. 12.3: Wege zum Geschlecht. Die Geschlechtsfestlegung über X- und Y-Chromosomen führt zu einem Gendosisproblem: Bei einem Geschlecht liegen doppelt so viele X-Allele vor, als bei dem anderen. Das Ausschalten eines X-Chromosoms oder Herunterfahren der Genaktivität auf beiden X-Chromosomen löst das Problem. Kapitel 13: Gentherapien
▼ ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ●
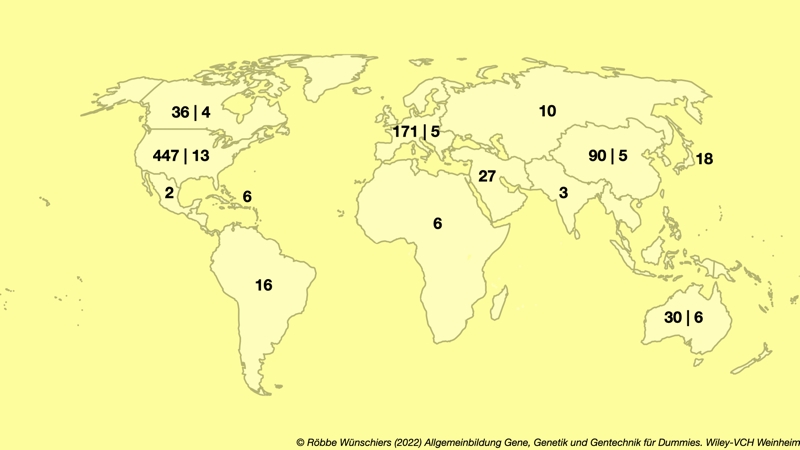
Abb. 13.1: Weltweit registrierte Gentherapien. Die Weltordnung auf meiner Karte ist etwas offener und auch Europa vereinter. Die Zahlen geben die Anzahl der beim US National Institutes of Health registrierten Gentherapie- und gegebenenfalls Geneditierungsstudien an, die sich in der klinischen Phase befinden: In Deutschland sind dies 38 Gentherapiestudien und eine Genditierungsstudie. Quelle: clinicaltrials.gov.
● ● ▼ ● ● ● ● ●
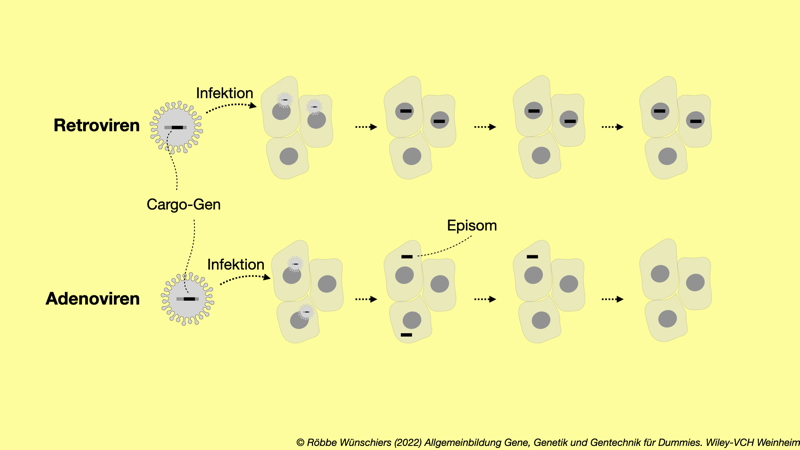
Abb. 13.2: Retro- und Adenoviren als Genfähren bei der Gentherapie. Retro- und Adenoviren können mit zusätzlicher genetischer Information (Cargo-Gen) ausgestattet werden. Bei der Infektion einer Wirtszelle bauen Retroviren ihr Erbgut mit dem Cargo-Gen stabil in das Wirtserbgut ein. Adenoviren und Adeno-assoziierte Viren (AAV) hinterlassen in der Wirtszelle ein sogenanntes Episom, das die genetische Information inklusive Cargo-Gen enthält. Episomen werden bei der Zellteilung nicht weitergegeben und gehen daher nach einiger Zeit verloren.
● ● ● ▼ ● ● ● ●
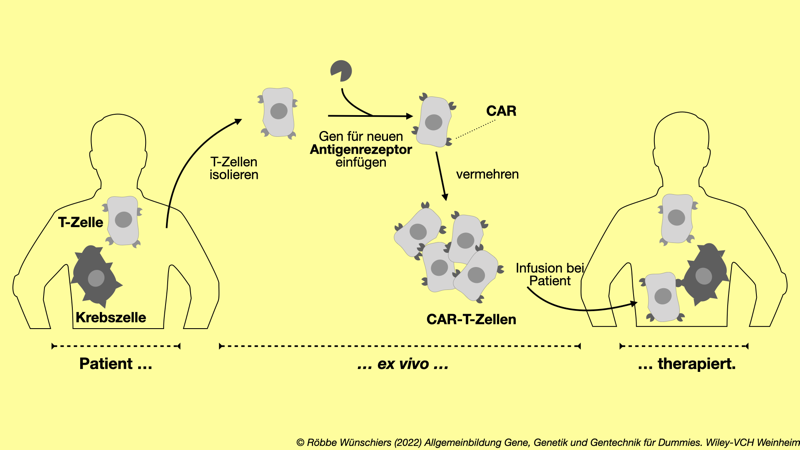
Abb. 13.3: Geneditierung bei der Immuntherapie gegen Krebs. T-Zellen des Immunsystems werden ex vivo gentechnisch verändert. Ihnen wird ein Genabschnitt für eine neue Erkennungsstelle des Antigenrezeptors eingesetzt, sodass ein chimärer Antigenrezeptor entsteht.
● ● ● ● ▼ ● ● ●
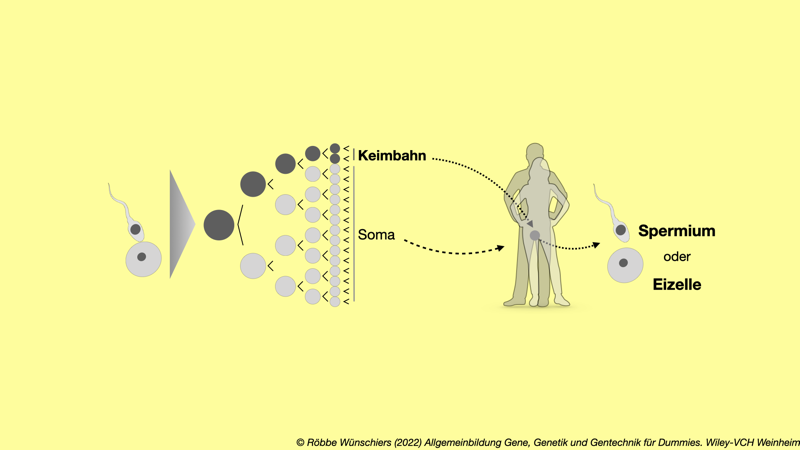
Abb. 13.4: Die Keimbahn. Die Zellen der Keimbahn entwickeln sich zu den Spermien und Eizellen, die bei der geschlechtlichen Fortpflanzung die nächste Generation begründen. Diese Zellen sind bei Säugetieren von den somatischen Zellen getrennt, welche die restlichen Körperzellen ausbilden.
● ● ● ● ● ▼ ● ●
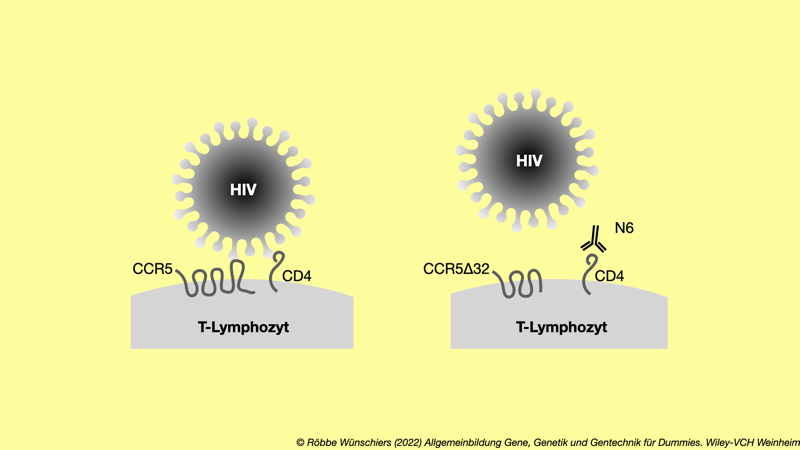
Abb. 13.5: Abwehr der HIV-Infektion einer weißen Blutzelle der Immunabwehr durch das CCR5-Delta-32-Allel oder den N6-Antikörper. Dazu muss er an den CCR5-Rezeptor und das CD4-Glykoprotein binden. Rechts) Das Fehlen von 32 Basenpaaren (Delta-32) im CCR5-Gen führt dazu, dass das Virus das Protein nicht mehr erkennt. Der gleiche Effekt kann mit dem Antikörper N6 erreicht werden, der an das Glykoprotein CD4 bindet und es damit »verdeckt«. Beide Mechanismen führen zu einer Immunität gegen HIV.
● ● ● ● ● ● ▼ ●
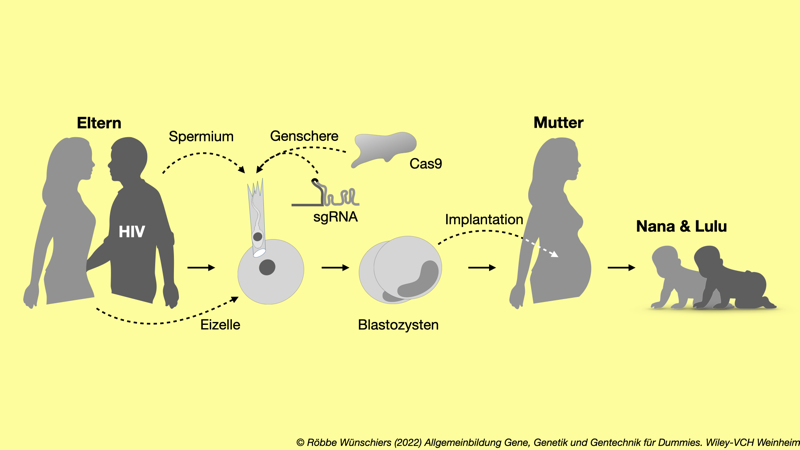
Abb. 13.6: Die Zwillinge Lulu und Nana sind die ersten geneditierten Babys. Während der In-vitro-Fertilisation beziehungsweise, genauer, der intrazytoplasmatischen Spermieninjektion (ICSI) wurde ihnen neben dem Spermium auch das Protein der DNA-Nuklease Cas9 und eine single guide RNA (sgRNA) injiziert. Cas9 und die sgRNA bilden gemeinsam die Genschere.
● ● ● ● ● ● ● ▼
Abb. 13.7: Erste Anwendung der Genschere CRISPR/Cas in der menschlichen Keimbahn. Das Ziel des Eingriffs war die Zerstörung der Funktion des CCR5-Rezeptor codierenden Gens auf Chromosom 3. Bei Lulu wurde nur in einem Allel eine fünfzehn Basenpaare Deletion erzielt. Dem resultierenden Protein fehlen intern fünf Aminosäuren. Bei Nana wurde eine vier Basenpaare große Deletion und eine ein Basenpaar große Insertion erreicht. Beides führt zu Leserasterverschiebungen und vorzeitigem Abbruch der Translation durch das Entstehen von Stopcodons. Kapitel 14: Wie Transhumanisten uns für die Zukunft wappnen (wollen)
▼
Kunst am Buch – von Kerstin Zentner. Kapitel 15: Coronavirus – Wenn »negativ« positiv ist
▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
Kunst am Buch – von Kerstin Zentner.
● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●
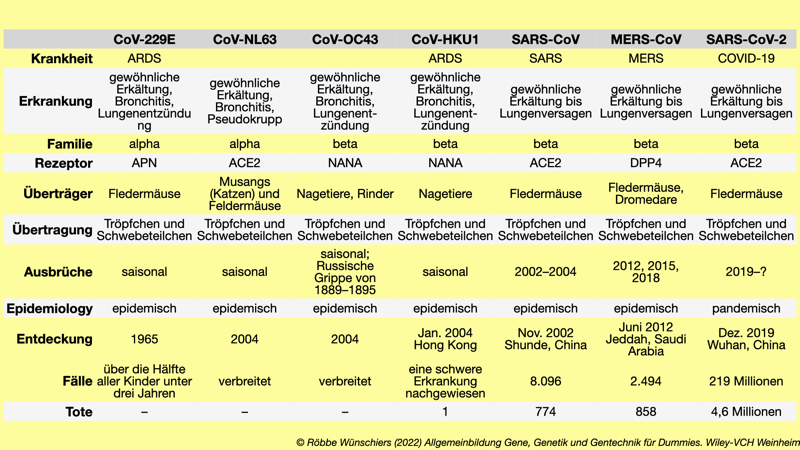
Tab. 15.1: Den Menschen infizierende Coronaviren. ARDS: acute respiratory distress syndrome, deutsch: Akutes Lungenversagen; NANA: N-Acetylneuraminic acid; DPP4: Dipeptidylpeptidase 4. Stand: September 2021.
● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ● ●
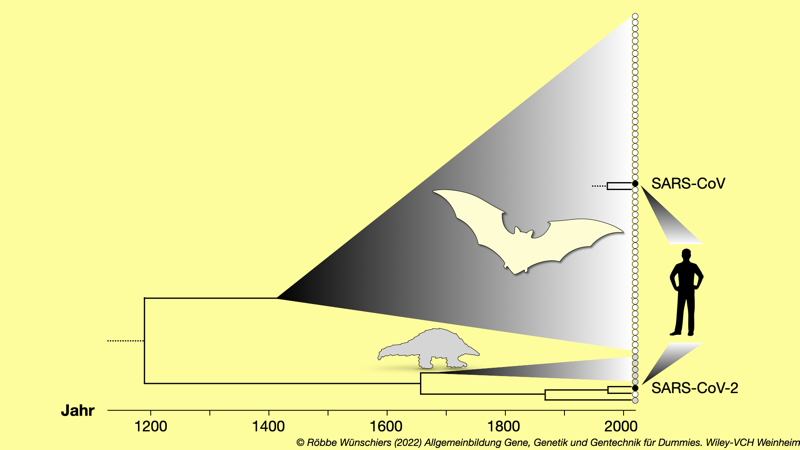
Abb. 15.1: Die Verwandtschaft von dreiundsechzig Coronaviren. Jeder Kreis am rechten Rand repräsentiert einen Coronavirusstamm. Weiße stehen für Virengenome, die aus Fledermäusen und graue Kreise für Coronaviren die aus Gürteltieren isoliert wurden. Schwarze Kreise repräsentieren SARS-CoV und SARS-CoV-2 aus menschlichen Proben. SARS-CoV und SARS-CoV-2 haben sich vor rund achthundert Jahren von ihrem nächsten Verwandten, jeweils einem Coronavirus aus der Fledermaus, getrennt.
● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ● ●
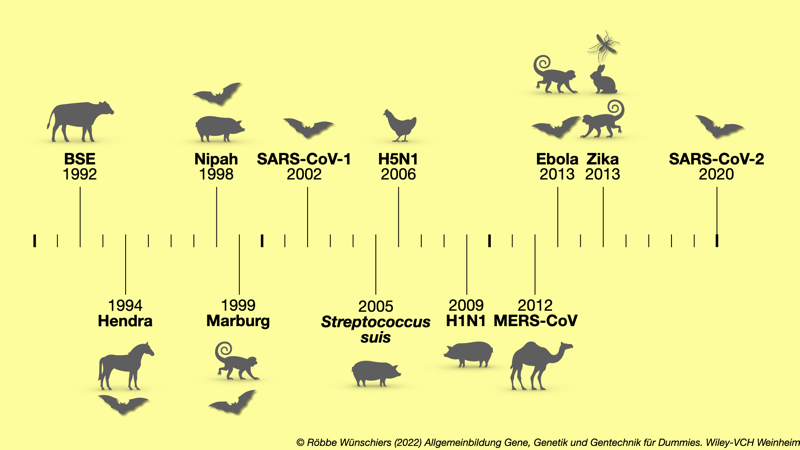
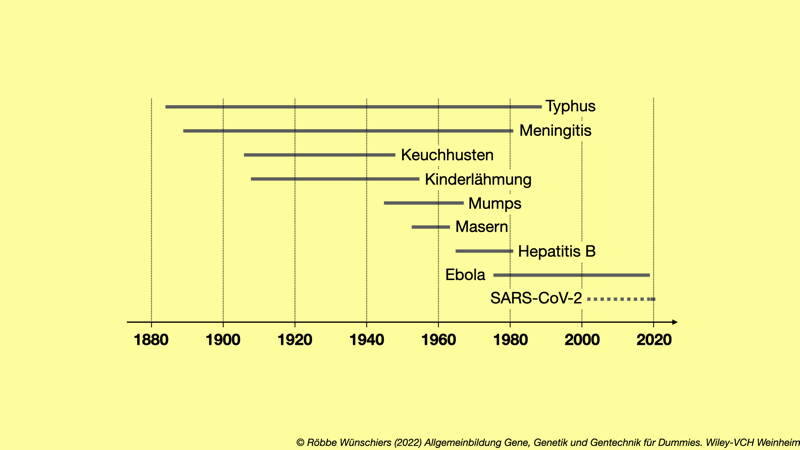
Abb. 15.2: Wesentliche Zoonosen der letzten dreißig Jahre. Gezeigt sind das Auftreten von Zoonosen und die wesentlichen Reservoirs beziehungsweise Vektoren.
● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ● ●
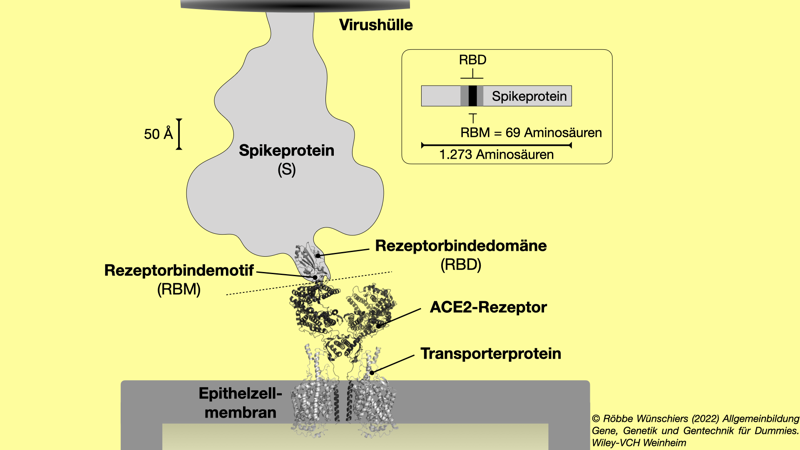
Abb. 15.3: Bindung von SARS-CoV-2 an eine Epithelzelle. Das Spikeprotein des Coronavirus hat ein Rezeptorbindemotif in der Rezeptorbindedomäne, die einen Teil des ACE2-Rezeptorproteins erkennt. Diese »Erkennung« entspricht letztlich einer molekularen Wechselwirkung auf der Ebene einiger Dutzend Aminosäuren. Führt die Wechselwirkung zu einer Bindung, initiiert dies das Eindringen des Virus in die Zelle. Oberhalb der gestrichelten Linie befindet sich das Virus, darunter die menschliche Zelle. Ein Ångström entspricht einer Strecke von 0,0001 Mikrometer. Molekulare Strukturen auf Basis der PDB-Daten 6M17 und 6VSB.
● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ● ●
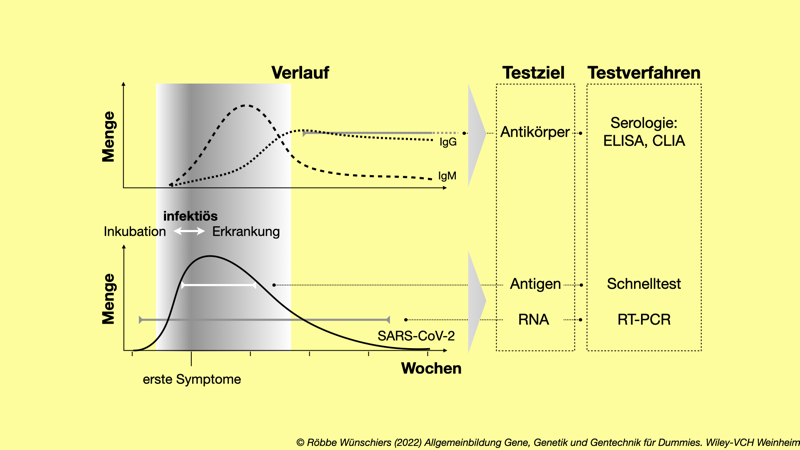
Abb. 15.4: Beispielhafter zeitlicher Verlauf und Nachweis einer SARS-CoV-2-Infektion. Der individuelle Verlauf kann abweichen, aber generell zeigen sich etwa eine Woche nach der Infektion erste Symptome. Unser Immunsystem reagiert auf die Infektion mit der Bildung von Proteinen, insbesondere den Immunglobulinen M und G (IgM, IgG). Gezeigt ist auch, in welchen Zeiträumen von den üblichen Testverfahren positive Ergebnisse zu erwarten sind. Die Testverfahren und was »Elisa« damit zu tun hat, erläutere ich im Text.
● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ● ●
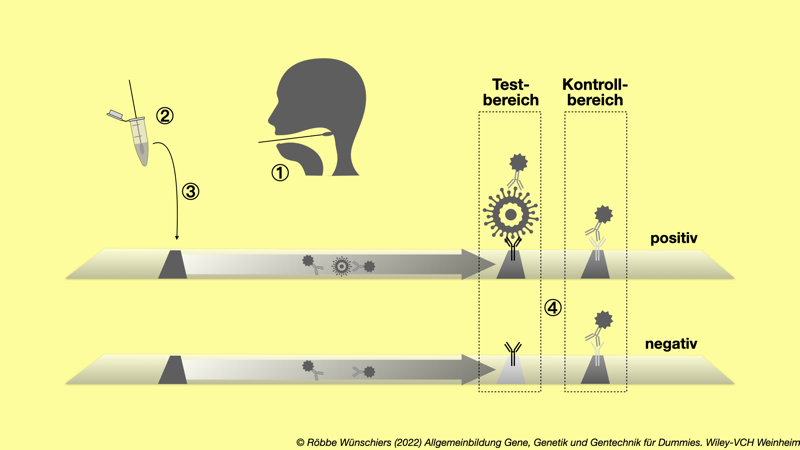
Abb. 15.5: Der Corona-Schnelltest ist ein Antigentest, beim das Virus direkt nachgewiesen wird. In der gleichen Weise funktioniert ein Schwangerschaftstest, wobei das nachzuweisende Antigen dabei natürlich kein Virus, sondern ein Hormon, das humane Choriongonadotropin, kurz hCG, ist.
● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ● ●
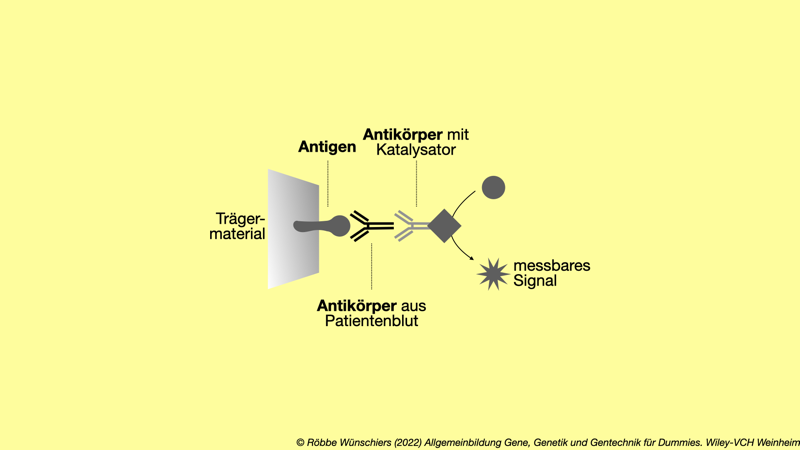
Abb. 15.6: Beim Immunassay wird getestet, ob der Patient Antikörper gegen ein Antigen im Blutserum trägt. Dazu wird das Blutserum mit einem immobilsierten Antigen in Verbindung gebracht. Nachdem alle nicht bindenden Antikörper aus dem Serum weggespült wurden, werden mit einem gegen menschliche Antikörper gerichteten universalen Antikörper (an den zusätzlich ein Katalysator, meist ein Enzym, gekoppelt ist) Antigen-bindende Patienten-Antikörper nachgewiesen.
● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ● ●
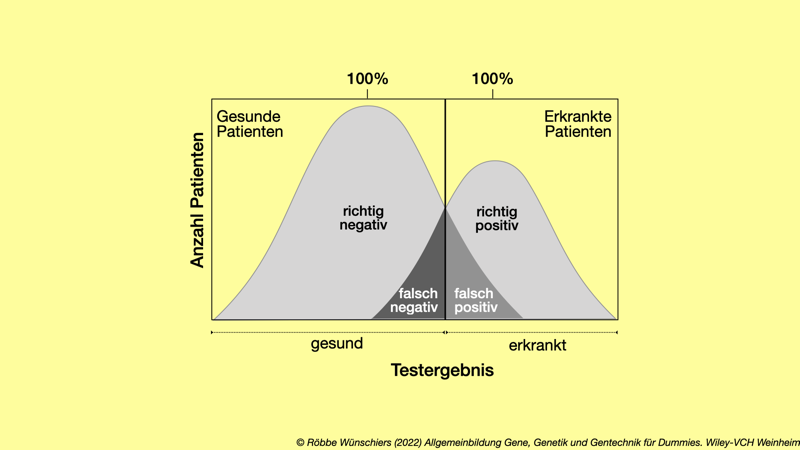
Abb. 15.7: Sensitivität und Spezifität sind wichtige Kriterien für diagnostische Tests. Selten liegen beide Werte bei hundert Prozent.
● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ● ●
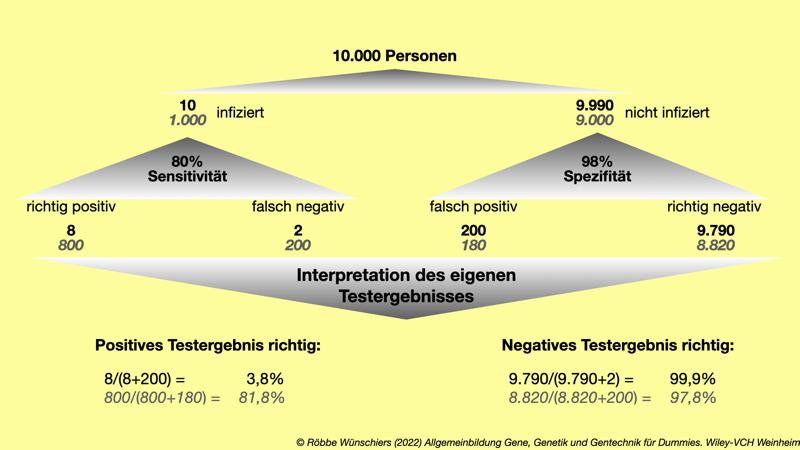
Abb. 15.8: Die Aussagekraft des eigenen Testergebnisses hängt von der Durchseuchung beziehungsweise der Teststrategie ab. Sind viele Infizierte unter den Testergebnissen (graue, kursive Zahlen), so steigt die Aussagekraft positiver Testergebnisse.
● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ● ●
Abb. 15.9: Impfstoffentwicklung. Der Zeitraum von der Detektion eines Erregers bis zu einem einsatzbereiten Impfstoff hat sich in den vergangen Jahrzehnten generell verkürzt. Die kürzeste Zeit benötigte die Impfstoffentwicklung gegen SARS-CoV-2. Allerdings konnte hier auf Erkenntnisse aus der SARS-CoV-1-Epidemie aufgebaut werden. Zudem hatten bei den Behörden die Prüfungs- und Genehmigungsverfahren Vorrang.
● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ● ●
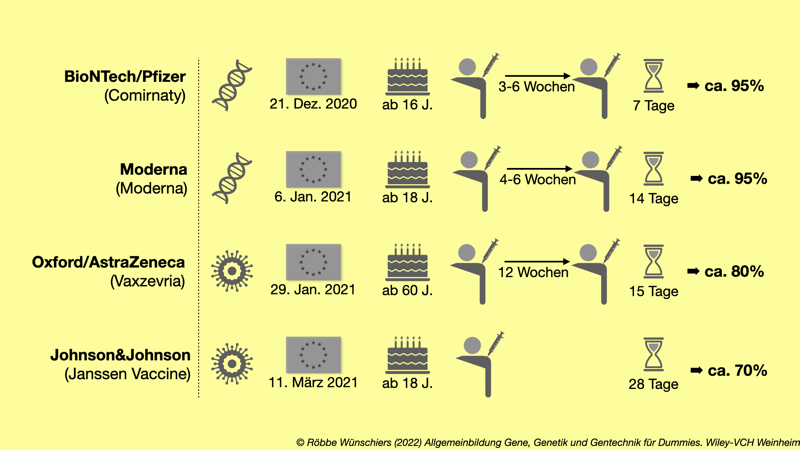
Abb. 15.10: Mit Stand vom Juni 2021 in der Europäischen Union zugelassene Impfstoffe gegen SARS-CoV-2. Gezeigt ist neben dem Impfstofftyp, der empfohlenen Altersgruppe und der Anzahl der notwendigen Dosen, die Zeit, bis die maximale Wirksamkeit erreicht ist.
● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ● ●
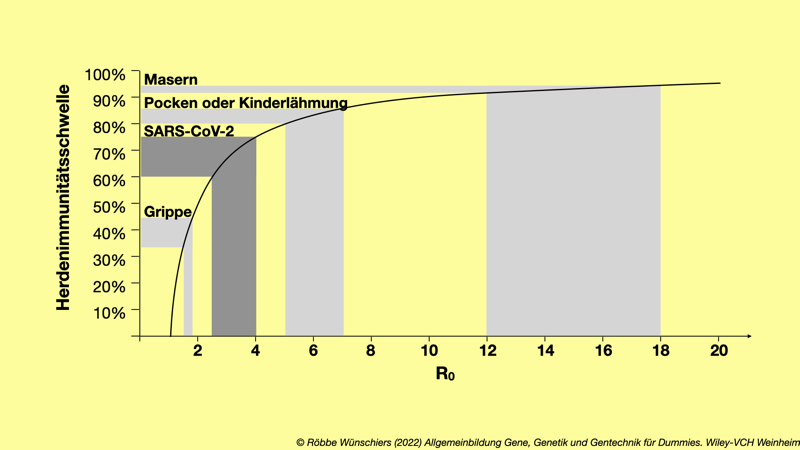
Abb. 15.11: Der Anteil der Bevölkerung der immunisiert sein muss, um Herdenimmunität zu erreichen, hängt von der Basisreproduktionszahl R$_{0}$ ab. Diese wird im Verlauf einer Epidemie bestimmt und kann sich durch das Auftreten von Mutanten auch verändern.
● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ● ●
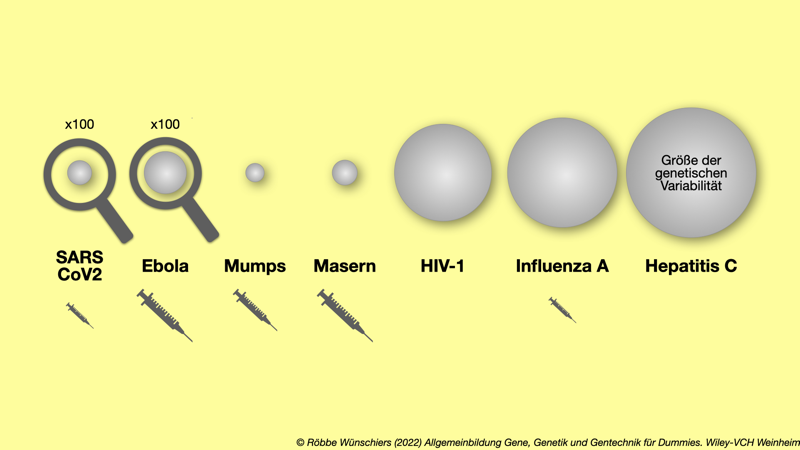
Abb. 15.12: Die genetische Variabilität verschiedener Viren und die Verfügbarkeit von Impfstoffen. Bei SARS-CoV-2 und Ebola ist die genetische Variabilität hundertfach vergrößert. Im Januar 2020 war die Variabilität von SARS-CoV-2 noch recht gering. Die große Sorge besteht darin, dass sie durch Rekombination rasant größer wird und das Ausmaß der Influenza A Viren erreicht. Hier kann nur mit großem Aufwand ein vergleichsweise schlecht wirkender Impfstoff bereitgestellt werden, der jährlich angepasst werden muss.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ● ●
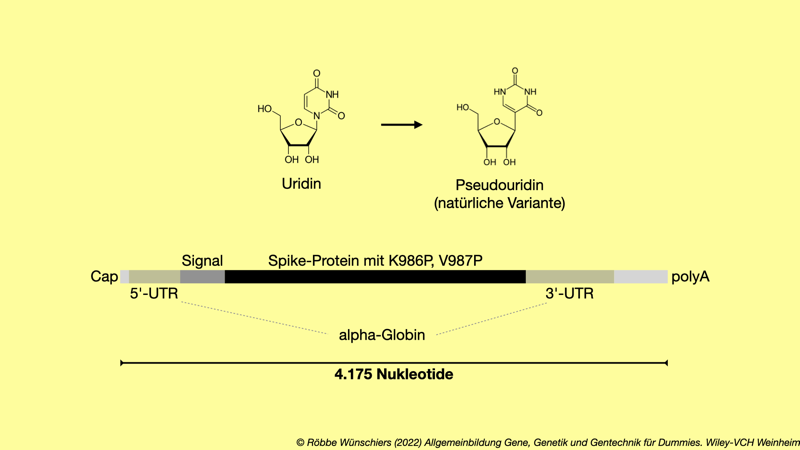
Abb. 15.13: Der SARS-CoV-2-Impfstoff der Firma BioNTech/Pfizer. Das Spikeprotein wurde an zwei Stellen verändert, um eine bessere Immunantwort zu erreichen: An Position 986 ist anstelle der Aminosäure Lysin (K) und an Position 987 anstelle der Aminosäure Valin (V) jeweils in ein Prolin (P) codiert. Das Ribonukleosid Uridin (Uracil) ist durchgängig durch die stabileren Variante Pseudouridin ersetzt. Ein Signalpeptid bewirkt den Transport des Spikeproteins aus der Oberarmmuskelzelle heraus, damit das Immunsystem das Spikeprotein auch zu sehen bekommt. Die nicht proteincodierenden flankierenden Bereiche stabilisieren die mRNA. Der Impfstoff von Moderna ist sehr ähnlich aufgebaut.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼ ●
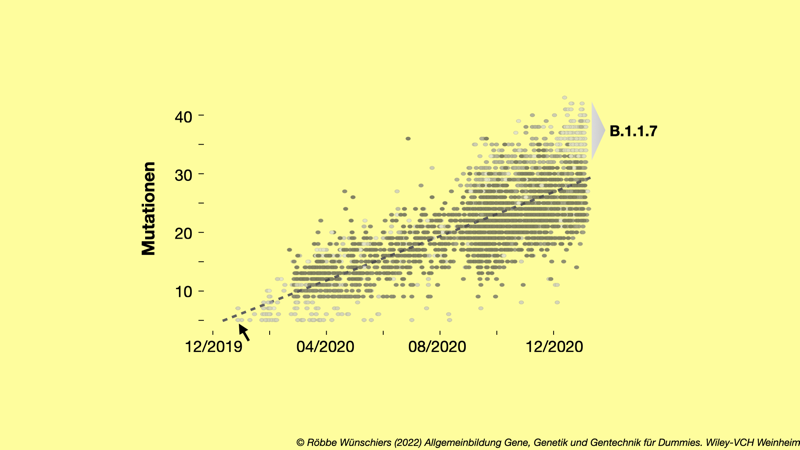
Abb. 15.14: Ansammlungen von Mutationen bei SARS-CoV-2 seit Beginn des Ausbruchs. Jeder der 3.915 Punkte repräsentiert ein SARS-CoV-2 Genom eines Patienten. Unterschiedliche Graustufen markieren einander ähnliche Varianten. Der Pfeil markiert den das SARS-CoV-2 Genom Wuhan/Hu-1/2019 des Patienten Null vom 29.~Dezember 2019. Die gestrichelte Linie zeigt die durchschnittliche Veränderung über der Zeit an. Zum Zeitpunkt der Erstellung dieser Grafik am 18. Januar 2021 sind dies 1,87 Mutationen pro Monat. Die Alpha-Variante B.1.1.7 mit erhöhter Infektiosität setzt sich oben rechts ab. Daten von nextstrain.org.
● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ▼
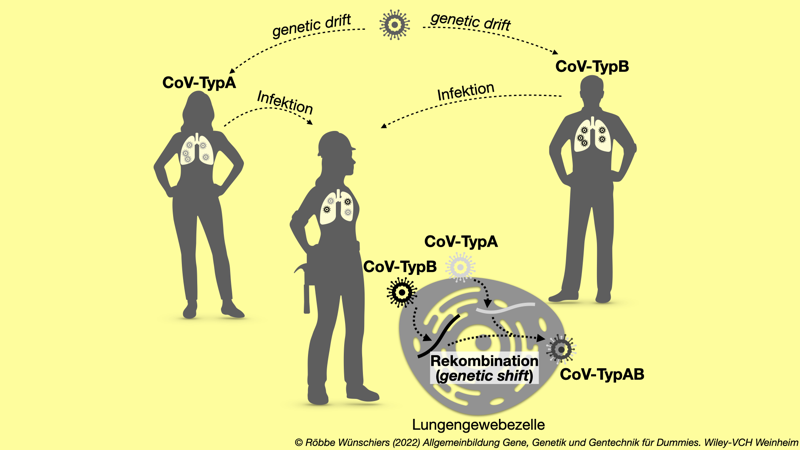
Abb. 15.15: Wirkung von Mutation und Rekombination. Ständig auftretende Mutationen verändern das Virus leicht und führen zum Genetic Shift. Infizieren zwei oder mehr genetisch unterschiedliche Virusvarianten dieselbe Zelle, kann es zum genetischen Austausch zwischen den Varianten kommen. Dieser Genetic Drift infolge von Rekombinationsereignissen führt zu größeren, oft sprunghaften Veränderungen. Kapitel 16: Zehn innovative Methoden im Bereich der Bio- und Gentechnologie
Dieses Kapitel hat keine Abbildungen – nur Methoden ;-)